Purdue Online Writing Lab Purdue OWL® College of Liberal Arts

Writing a Literature Review

Welcome to the Purdue OWL
This page is brought to you by the OWL at Purdue University. When printing this page, you must include the entire legal notice.
Copyright ©1995-2018 by The Writing Lab & The OWL at Purdue and Purdue University. All rights reserved. This material may not be published, reproduced, broadcast, rewritten, or redistributed without permission. Use of this site constitutes acceptance of our terms and conditions of fair use.
A literature review is a document or section of a document that collects key sources on a topic and discusses those sources in conversation with each other (also called synthesis ). The lit review is an important genre in many disciplines, not just literature (i.e., the study of works of literature such as novels and plays). When we say “literature review” or refer to “the literature,” we are talking about the research ( scholarship ) in a given field. You will often see the terms “the research,” “the scholarship,” and “the literature” used mostly interchangeably.
Where, when, and why would I write a lit review?
There are a number of different situations where you might write a literature review, each with slightly different expectations; different disciplines, too, have field-specific expectations for what a literature review is and does. For instance, in the humanities, authors might include more overt argumentation and interpretation of source material in their literature reviews, whereas in the sciences, authors are more likely to report study designs and results in their literature reviews; these differences reflect these disciplines’ purposes and conventions in scholarship. You should always look at examples from your own discipline and talk to professors or mentors in your field to be sure you understand your discipline’s conventions, for literature reviews as well as for any other genre.
A literature review can be a part of a research paper or scholarly article, usually falling after the introduction and before the research methods sections. In these cases, the lit review just needs to cover scholarship that is important to the issue you are writing about; sometimes it will also cover key sources that informed your research methodology.
Lit reviews can also be standalone pieces, either as assignments in a class or as publications. In a class, a lit review may be assigned to help students familiarize themselves with a topic and with scholarship in their field, get an idea of the other researchers working on the topic they’re interested in, find gaps in existing research in order to propose new projects, and/or develop a theoretical framework and methodology for later research. As a publication, a lit review usually is meant to help make other scholars’ lives easier by collecting and summarizing, synthesizing, and analyzing existing research on a topic. This can be especially helpful for students or scholars getting into a new research area, or for directing an entire community of scholars toward questions that have not yet been answered.
What are the parts of a lit review?
Most lit reviews use a basic introduction-body-conclusion structure; if your lit review is part of a larger paper, the introduction and conclusion pieces may be just a few sentences while you focus most of your attention on the body. If your lit review is a standalone piece, the introduction and conclusion take up more space and give you a place to discuss your goals, research methods, and conclusions separately from where you discuss the literature itself.
Introduction:
- An introductory paragraph that explains what your working topic and thesis is
- A forecast of key topics or texts that will appear in the review
- Potentially, a description of how you found sources and how you analyzed them for inclusion and discussion in the review (more often found in published, standalone literature reviews than in lit review sections in an article or research paper)
- Summarize and synthesize: Give an overview of the main points of each source and combine them into a coherent whole
- Analyze and interpret: Don’t just paraphrase other researchers – add your own interpretations where possible, discussing the significance of findings in relation to the literature as a whole
- Critically Evaluate: Mention the strengths and weaknesses of your sources
- Write in well-structured paragraphs: Use transition words and topic sentence to draw connections, comparisons, and contrasts.
Conclusion:
- Summarize the key findings you have taken from the literature and emphasize their significance
- Connect it back to your primary research question
How should I organize my lit review?
Lit reviews can take many different organizational patterns depending on what you are trying to accomplish with the review. Here are some examples:
- Chronological : The simplest approach is to trace the development of the topic over time, which helps familiarize the audience with the topic (for instance if you are introducing something that is not commonly known in your field). If you choose this strategy, be careful to avoid simply listing and summarizing sources in order. Try to analyze the patterns, turning points, and key debates that have shaped the direction of the field. Give your interpretation of how and why certain developments occurred (as mentioned previously, this may not be appropriate in your discipline — check with a teacher or mentor if you’re unsure).
- Thematic : If you have found some recurring central themes that you will continue working with throughout your piece, you can organize your literature review into subsections that address different aspects of the topic. For example, if you are reviewing literature about women and religion, key themes can include the role of women in churches and the religious attitude towards women.
- Qualitative versus quantitative research
- Empirical versus theoretical scholarship
- Divide the research by sociological, historical, or cultural sources
- Theoretical : In many humanities articles, the literature review is the foundation for the theoretical framework. You can use it to discuss various theories, models, and definitions of key concepts. You can argue for the relevance of a specific theoretical approach or combine various theorical concepts to create a framework for your research.
What are some strategies or tips I can use while writing my lit review?
Any lit review is only as good as the research it discusses; make sure your sources are well-chosen and your research is thorough. Don’t be afraid to do more research if you discover a new thread as you’re writing. More info on the research process is available in our "Conducting Research" resources .
As you’re doing your research, create an annotated bibliography ( see our page on the this type of document ). Much of the information used in an annotated bibliography can be used also in a literature review, so you’ll be not only partially drafting your lit review as you research, but also developing your sense of the larger conversation going on among scholars, professionals, and any other stakeholders in your topic.
Usually you will need to synthesize research rather than just summarizing it. This means drawing connections between sources to create a picture of the scholarly conversation on a topic over time. Many student writers struggle to synthesize because they feel they don’t have anything to add to the scholars they are citing; here are some strategies to help you:
- It often helps to remember that the point of these kinds of syntheses is to show your readers how you understand your research, to help them read the rest of your paper.
- Writing teachers often say synthesis is like hosting a dinner party: imagine all your sources are together in a room, discussing your topic. What are they saying to each other?
- Look at the in-text citations in each paragraph. Are you citing just one source for each paragraph? This usually indicates summary only. When you have multiple sources cited in a paragraph, you are more likely to be synthesizing them (not always, but often
- Read more about synthesis here.
The most interesting literature reviews are often written as arguments (again, as mentioned at the beginning of the page, this is discipline-specific and doesn’t work for all situations). Often, the literature review is where you can establish your research as filling a particular gap or as relevant in a particular way. You have some chance to do this in your introduction in an article, but the literature review section gives a more extended opportunity to establish the conversation in the way you would like your readers to see it. You can choose the intellectual lineage you would like to be part of and whose definitions matter most to your thinking (mostly humanities-specific, but this goes for sciences as well). In addressing these points, you argue for your place in the conversation, which tends to make the lit review more compelling than a simple reporting of other sources.
Have a language expert improve your writing
Run a free plagiarism check in 10 minutes, generate accurate citations for free.
- Knowledge Base
Methodology
- How to Write a Literature Review | Guide, Examples, & Templates
How to Write a Literature Review | Guide, Examples, & Templates
Published on January 2, 2023 by Shona McCombes . Revised on September 11, 2023.
What is a literature review? A literature review is a survey of scholarly sources on a specific topic. It provides an overview of current knowledge, allowing you to identify relevant theories, methods, and gaps in the existing research that you can later apply to your paper, thesis, or dissertation topic .
There are five key steps to writing a literature review:
- Search for relevant literature
- Evaluate sources
- Identify themes, debates, and gaps
- Outline the structure
- Write your literature review
A good literature review doesn’t just summarize sources—it analyzes, synthesizes , and critically evaluates to give a clear picture of the state of knowledge on the subject.
Instantly correct all language mistakes in your text
Upload your document to correct all your mistakes in minutes

Table of contents
What is the purpose of a literature review, examples of literature reviews, step 1 – search for relevant literature, step 2 – evaluate and select sources, step 3 – identify themes, debates, and gaps, step 4 – outline your literature review’s structure, step 5 – write your literature review, free lecture slides, other interesting articles, frequently asked questions, introduction.
- Quick Run-through
- Step 1 & 2
When you write a thesis , dissertation , or research paper , you will likely have to conduct a literature review to situate your research within existing knowledge. The literature review gives you a chance to:
- Demonstrate your familiarity with the topic and its scholarly context
- Develop a theoretical framework and methodology for your research
- Position your work in relation to other researchers and theorists
- Show how your research addresses a gap or contributes to a debate
- Evaluate the current state of research and demonstrate your knowledge of the scholarly debates around your topic.
Writing literature reviews is a particularly important skill if you want to apply for graduate school or pursue a career in research. We’ve written a step-by-step guide that you can follow below.

Receive feedback on language, structure, and formatting
Professional editors proofread and edit your paper by focusing on:
- Academic style
- Vague sentences
- Style consistency
See an example

Writing literature reviews can be quite challenging! A good starting point could be to look at some examples, depending on what kind of literature review you’d like to write.
- Example literature review #1: “Why Do People Migrate? A Review of the Theoretical Literature” ( Theoretical literature review about the development of economic migration theory from the 1950s to today.)
- Example literature review #2: “Literature review as a research methodology: An overview and guidelines” ( Methodological literature review about interdisciplinary knowledge acquisition and production.)
- Example literature review #3: “The Use of Technology in English Language Learning: A Literature Review” ( Thematic literature review about the effects of technology on language acquisition.)
- Example literature review #4: “Learners’ Listening Comprehension Difficulties in English Language Learning: A Literature Review” ( Chronological literature review about how the concept of listening skills has changed over time.)
You can also check out our templates with literature review examples and sample outlines at the links below.
Download Word doc Download Google doc
Before you begin searching for literature, you need a clearly defined topic .
If you are writing the literature review section of a dissertation or research paper, you will search for literature related to your research problem and questions .
Make a list of keywords
Start by creating a list of keywords related to your research question. Include each of the key concepts or variables you’re interested in, and list any synonyms and related terms. You can add to this list as you discover new keywords in the process of your literature search.
- Social media, Facebook, Instagram, Twitter, Snapchat, TikTok
- Body image, self-perception, self-esteem, mental health
- Generation Z, teenagers, adolescents, youth
Search for relevant sources
Use your keywords to begin searching for sources. Some useful databases to search for journals and articles include:
- Your university’s library catalogue
- Google Scholar
- Project Muse (humanities and social sciences)
- Medline (life sciences and biomedicine)
- EconLit (economics)
- Inspec (physics, engineering and computer science)
You can also use boolean operators to help narrow down your search.
Make sure to read the abstract to find out whether an article is relevant to your question. When you find a useful book or article, you can check the bibliography to find other relevant sources.
You likely won’t be able to read absolutely everything that has been written on your topic, so it will be necessary to evaluate which sources are most relevant to your research question.
For each publication, ask yourself:
- What question or problem is the author addressing?
- What are the key concepts and how are they defined?
- What are the key theories, models, and methods?
- Does the research use established frameworks or take an innovative approach?
- What are the results and conclusions of the study?
- How does the publication relate to other literature in the field? Does it confirm, add to, or challenge established knowledge?
- What are the strengths and weaknesses of the research?
Make sure the sources you use are credible , and make sure you read any landmark studies and major theories in your field of research.
You can use our template to summarize and evaluate sources you’re thinking about using. Click on either button below to download.
Take notes and cite your sources
As you read, you should also begin the writing process. Take notes that you can later incorporate into the text of your literature review.
It is important to keep track of your sources with citations to avoid plagiarism . It can be helpful to make an annotated bibliography , where you compile full citation information and write a paragraph of summary and analysis for each source. This helps you remember what you read and saves time later in the process.
Don't submit your assignments before you do this
The academic proofreading tool has been trained on 1000s of academic texts. Making it the most accurate and reliable proofreading tool for students. Free citation check included.

Try for free
To begin organizing your literature review’s argument and structure, be sure you understand the connections and relationships between the sources you’ve read. Based on your reading and notes, you can look for:
- Trends and patterns (in theory, method or results): do certain approaches become more or less popular over time?
- Themes: what questions or concepts recur across the literature?
- Debates, conflicts and contradictions: where do sources disagree?
- Pivotal publications: are there any influential theories or studies that changed the direction of the field?
- Gaps: what is missing from the literature? Are there weaknesses that need to be addressed?
This step will help you work out the structure of your literature review and (if applicable) show how your own research will contribute to existing knowledge.
- Most research has focused on young women.
- There is an increasing interest in the visual aspects of social media.
- But there is still a lack of robust research on highly visual platforms like Instagram and Snapchat—this is a gap that you could address in your own research.
There are various approaches to organizing the body of a literature review. Depending on the length of your literature review, you can combine several of these strategies (for example, your overall structure might be thematic, but each theme is discussed chronologically).
Chronological
The simplest approach is to trace the development of the topic over time. However, if you choose this strategy, be careful to avoid simply listing and summarizing sources in order.
Try to analyze patterns, turning points and key debates that have shaped the direction of the field. Give your interpretation of how and why certain developments occurred.
If you have found some recurring central themes, you can organize your literature review into subsections that address different aspects of the topic.
For example, if you are reviewing literature about inequalities in migrant health outcomes, key themes might include healthcare policy, language barriers, cultural attitudes, legal status, and economic access.
Methodological
If you draw your sources from different disciplines or fields that use a variety of research methods , you might want to compare the results and conclusions that emerge from different approaches. For example:
- Look at what results have emerged in qualitative versus quantitative research
- Discuss how the topic has been approached by empirical versus theoretical scholarship
- Divide the literature into sociological, historical, and cultural sources
Theoretical
A literature review is often the foundation for a theoretical framework . You can use it to discuss various theories, models, and definitions of key concepts.
You might argue for the relevance of a specific theoretical approach, or combine various theoretical concepts to create a framework for your research.
Like any other academic text , your literature review should have an introduction , a main body, and a conclusion . What you include in each depends on the objective of your literature review.
The introduction should clearly establish the focus and purpose of the literature review.
Depending on the length of your literature review, you might want to divide the body into subsections. You can use a subheading for each theme, time period, or methodological approach.
As you write, you can follow these tips:
- Summarize and synthesize: give an overview of the main points of each source and combine them into a coherent whole
- Analyze and interpret: don’t just paraphrase other researchers — add your own interpretations where possible, discussing the significance of findings in relation to the literature as a whole
- Critically evaluate: mention the strengths and weaknesses of your sources
- Write in well-structured paragraphs: use transition words and topic sentences to draw connections, comparisons and contrasts
In the conclusion, you should summarize the key findings you have taken from the literature and emphasize their significance.
When you’ve finished writing and revising your literature review, don’t forget to proofread thoroughly before submitting. Not a language expert? Check out Scribbr’s professional proofreading services !
This article has been adapted into lecture slides that you can use to teach your students about writing a literature review.
Scribbr slides are free to use, customize, and distribute for educational purposes.
Open Google Slides Download PowerPoint
If you want to know more about the research process , methodology , research bias , or statistics , make sure to check out some of our other articles with explanations and examples.
- Sampling methods
- Simple random sampling
- Stratified sampling
- Cluster sampling
- Likert scales
- Reproducibility
Statistics
- Null hypothesis
- Statistical power
- Probability distribution
- Effect size
- Poisson distribution
Research bias
- Optimism bias
- Cognitive bias
- Implicit bias
- Hawthorne effect
- Anchoring bias
- Explicit bias
A literature review is a survey of scholarly sources (such as books, journal articles, and theses) related to a specific topic or research question .
It is often written as part of a thesis, dissertation , or research paper , in order to situate your work in relation to existing knowledge.
There are several reasons to conduct a literature review at the beginning of a research project:
- To familiarize yourself with the current state of knowledge on your topic
- To ensure that you’re not just repeating what others have already done
- To identify gaps in knowledge and unresolved problems that your research can address
- To develop your theoretical framework and methodology
- To provide an overview of the key findings and debates on the topic
Writing the literature review shows your reader how your work relates to existing research and what new insights it will contribute.
The literature review usually comes near the beginning of your thesis or dissertation . After the introduction , it grounds your research in a scholarly field and leads directly to your theoretical framework or methodology .
A literature review is a survey of credible sources on a topic, often used in dissertations , theses, and research papers . Literature reviews give an overview of knowledge on a subject, helping you identify relevant theories and methods, as well as gaps in existing research. Literature reviews are set up similarly to other academic texts , with an introduction , a main body, and a conclusion .
An annotated bibliography is a list of source references that has a short description (called an annotation ) for each of the sources. It is often assigned as part of the research process for a paper .
Cite this Scribbr article
If you want to cite this source, you can copy and paste the citation or click the “Cite this Scribbr article” button to automatically add the citation to our free Citation Generator.
McCombes, S. (2023, September 11). How to Write a Literature Review | Guide, Examples, & Templates. Scribbr. Retrieved August 12, 2024, from https://www.scribbr.com/dissertation/literature-review/
Is this article helpful?

Shona McCombes
Other students also liked, what is a theoretical framework | guide to organizing, what is a research methodology | steps & tips, how to write a research proposal | examples & templates, get unlimited documents corrected.
✔ Free APA citation check included ✔ Unlimited document corrections ✔ Specialized in correcting academic texts
Research Methods
- Getting Started
- Literature Review Research
- Research Design
- Research Design By Discipline
- SAGE Research Methods
- Teaching with SAGE Research Methods
Literature Review
- What is a Literature Review?
- What is NOT a Literature Review?
- Purposes of a Literature Review
- Types of Literature Reviews
- Literature Reviews vs. Systematic Reviews
- Systematic vs. Meta-Analysis
Literature Review is a comprehensive survey of the works published in a particular field of study or line of research, usually over a specific period of time, in the form of an in-depth, critical bibliographic essay or annotated list in which attention is drawn to the most significant works.
Also, we can define a literature review as the collected body of scholarly works related to a topic:
- Summarizes and analyzes previous research relevant to a topic
- Includes scholarly books and articles published in academic journals
- Can be an specific scholarly paper or a section in a research paper
The objective of a Literature Review is to find previous published scholarly works relevant to an specific topic
- Help gather ideas or information
- Keep up to date in current trends and findings
- Help develop new questions
A literature review is important because it:
- Explains the background of research on a topic.
- Demonstrates why a topic is significant to a subject area.
- Helps focus your own research questions or problems
- Discovers relationships between research studies/ideas.
- Suggests unexplored ideas or populations
- Identifies major themes, concepts, and researchers on a topic.
- Tests assumptions; may help counter preconceived ideas and remove unconscious bias.
- Identifies critical gaps, points of disagreement, or potentially flawed methodology or theoretical approaches.
- Indicates potential directions for future research.
All content in this section is from Literature Review Research from Old Dominion University
Keep in mind the following, a literature review is NOT:
Not an essay
Not an annotated bibliography in which you summarize each article that you have reviewed. A literature review goes beyond basic summarizing to focus on the critical analysis of the reviewed works and their relationship to your research question.
Not a research paper where you select resources to support one side of an issue versus another. A lit review should explain and consider all sides of an argument in order to avoid bias, and areas of agreement and disagreement should be highlighted.
A literature review serves several purposes. For example, it
- provides thorough knowledge of previous studies; introduces seminal works.
- helps focus one’s own research topic.
- identifies a conceptual framework for one’s own research questions or problems; indicates potential directions for future research.
- suggests previously unused or underused methodologies, designs, quantitative and qualitative strategies.
- identifies gaps in previous studies; identifies flawed methodologies and/or theoretical approaches; avoids replication of mistakes.
- helps the researcher avoid repetition of earlier research.
- suggests unexplored populations.
- determines whether past studies agree or disagree; identifies controversy in the literature.
- tests assumptions; may help counter preconceived ideas and remove unconscious bias.
As Kennedy (2007) notes*, it is important to think of knowledge in a given field as consisting of three layers. First, there are the primary studies that researchers conduct and publish. Second are the reviews of those studies that summarize and offer new interpretations built from and often extending beyond the original studies. Third, there are the perceptions, conclusions, opinion, and interpretations that are shared informally that become part of the lore of field. In composing a literature review, it is important to note that it is often this third layer of knowledge that is cited as "true" even though it often has only a loose relationship to the primary studies and secondary literature reviews.
Given this, while literature reviews are designed to provide an overview and synthesis of pertinent sources you have explored, there are several approaches to how they can be done, depending upon the type of analysis underpinning your study. Listed below are definitions of types of literature reviews:
Argumentative Review This form examines literature selectively in order to support or refute an argument, deeply imbedded assumption, or philosophical problem already established in the literature. The purpose is to develop a body of literature that establishes a contrarian viewpoint. Given the value-laden nature of some social science research [e.g., educational reform; immigration control], argumentative approaches to analyzing the literature can be a legitimate and important form of discourse. However, note that they can also introduce problems of bias when they are used to to make summary claims of the sort found in systematic reviews.
Integrative Review Considered a form of research that reviews, critiques, and synthesizes representative literature on a topic in an integrated way such that new frameworks and perspectives on the topic are generated. The body of literature includes all studies that address related or identical hypotheses. A well-done integrative review meets the same standards as primary research in regard to clarity, rigor, and replication.
Historical Review Few things rest in isolation from historical precedent. Historical reviews are focused on examining research throughout a period of time, often starting with the first time an issue, concept, theory, phenomena emerged in the literature, then tracing its evolution within the scholarship of a discipline. The purpose is to place research in a historical context to show familiarity with state-of-the-art developments and to identify the likely directions for future research.
Methodological Review A review does not always focus on what someone said [content], but how they said it [method of analysis]. This approach provides a framework of understanding at different levels (i.e. those of theory, substantive fields, research approaches and data collection and analysis techniques), enables researchers to draw on a wide variety of knowledge ranging from the conceptual level to practical documents for use in fieldwork in the areas of ontological and epistemological consideration, quantitative and qualitative integration, sampling, interviewing, data collection and data analysis, and helps highlight many ethical issues which we should be aware of and consider as we go through our study.
Systematic Review This form consists of an overview of existing evidence pertinent to a clearly formulated research question, which uses pre-specified and standardized methods to identify and critically appraise relevant research, and to collect, report, and analyse data from the studies that are included in the review. Typically it focuses on a very specific empirical question, often posed in a cause-and-effect form, such as "To what extent does A contribute to B?"
Theoretical Review The purpose of this form is to concretely examine the corpus of theory that has accumulated in regard to an issue, concept, theory, phenomena. The theoretical literature review help establish what theories already exist, the relationships between them, to what degree the existing theories have been investigated, and to develop new hypotheses to be tested. Often this form is used to help establish a lack of appropriate theories or reveal that current theories are inadequate for explaining new or emerging research problems. The unit of analysis can focus on a theoretical concept or a whole theory or framework.
* Kennedy, Mary M. "Defining a Literature." Educational Researcher 36 (April 2007): 139-147.
All content in this section is from The Literature Review created by Dr. Robert Larabee USC
Robinson, P. and Lowe, J. (2015), Literature reviews vs systematic reviews. Australian and New Zealand Journal of Public Health, 39: 103-103. doi: 10.1111/1753-6405.12393

What's in the name? The difference between a Systematic Review and a Literature Review, and why it matters . By Lynn Kysh from University of Southern California

Systematic review or meta-analysis?
A systematic review answers a defined research question by collecting and summarizing all empirical evidence that fits pre-specified eligibility criteria.
A meta-analysis is the use of statistical methods to summarize the results of these studies.
Systematic reviews, just like other research articles, can be of varying quality. They are a significant piece of work (the Centre for Reviews and Dissemination at York estimates that a team will take 9-24 months), and to be useful to other researchers and practitioners they should have:
- clearly stated objectives with pre-defined eligibility criteria for studies
- explicit, reproducible methodology
- a systematic search that attempts to identify all studies
- assessment of the validity of the findings of the included studies (e.g. risk of bias)
- systematic presentation, and synthesis, of the characteristics and findings of the included studies
Not all systematic reviews contain meta-analysis.
Meta-analysis is the use of statistical methods to summarize the results of independent studies. By combining information from all relevant studies, meta-analysis can provide more precise estimates of the effects of health care than those derived from the individual studies included within a review. More information on meta-analyses can be found in Cochrane Handbook, Chapter 9 .
A meta-analysis goes beyond critique and integration and conducts secondary statistical analysis on the outcomes of similar studies. It is a systematic review that uses quantitative methods to synthesize and summarize the results.
An advantage of a meta-analysis is the ability to be completely objective in evaluating research findings. Not all topics, however, have sufficient research evidence to allow a meta-analysis to be conducted. In that case, an integrative review is an appropriate strategy.
Some of the content in this section is from Systematic reviews and meta-analyses: step by step guide created by Kate McAllister.
- << Previous: Getting Started
- Next: Research Design >>
- Last Updated: Jul 15, 2024 10:34 AM
- URL: https://guides.lib.udel.edu/researchmethods
Libraries | Research Guides
Literature reviews, what is a literature review, learning more about how to do a literature review.
- Planning the Review
- The Research Question
- Choosing Where to Search
- Organizing the Review
- Writing the Review
A literature review is a review and synthesis of existing research on a topic or research question. A literature review is meant to analyze the scholarly literature, make connections across writings and identify strengths, weaknesses, trends, and missing conversations. A literature review should address different aspects of a topic as it relates to your research question. A literature review goes beyond a description or summary of the literature you have read.
- Sage Research Methods Core This link opens in a new window SAGE Research Methods supports research at all levels by providing material to guide users through every step of the research process. SAGE Research Methods is the ultimate methods library with more than 1000 books, reference works, journal articles, and instructional videos by world-leading academics from across the social sciences, including the largest collection of qualitative methods books available online from any scholarly publisher. – Publisher

- Next: Planning the Review >>
- Last Updated: Jul 8, 2024 11:22 AM
- URL: https://libguides.northwestern.edu/literaturereviews

Research Methods: Literature Reviews
- Annotated Bibliographies
- Literature Reviews
- Scoping Reviews
- Systematic Reviews
- Scholarship of Teaching and Learning
- Persuasive Arguments
- Subject Specific Methodology
A literature review involves researching, reading, analyzing, evaluating, and summarizing scholarly literature (typically journals and articles) about a specific topic. The results of a literature review may be an entire report or article OR may be part of a article, thesis, dissertation, or grant proposal. A literature review helps the author learn about the history and nature of their topic, and identify research gaps and problems.
Steps & Elements
Problem formulation
- Determine your topic and its components by asking a question
- Research: locate literature related to your topic to identify the gap(s) that can be addressed
- Read: read the articles or other sources of information
- Analyze: assess the findings for relevancy
- Evaluating: determine how the article are relevant to your research and what are the key findings
- Synthesis: write about the key findings and how it is relevant to your research
Elements of a Literature Review
- Summarize subject, issue or theory under consideration, along with objectives of the review
- Divide works under review into categories (e.g. those in support of a particular position, those against, those offering alternative theories entirely)
- Explain how each work is similar to and how it varies from the others
- Conclude which pieces are best considered in their argument, are most convincing of their opinions, and make the greatest contribution to the understanding and development of an area of research
Writing a Literature Review Resources
- How to Write a Literature Review From the Wesleyan University Library
- Write a Literature Review From the University of California Santa Cruz Library. A Brief overview of a literature review, includes a list of stages for writing a lit review.
- Literature Reviews From the University of North Carolina Writing Center. Detailed information about writing a literature review.
- Undertaking a literature review: a step-by-step approach Cronin, P., Ryan, F., & Coughan, M. (2008). Undertaking a literature review: A step-by-step approach. British Journal of Nursing, 17(1), p.38-43

Literature Review Tutorial
- << Previous: Annotated Bibliographies
- Next: Scoping Reviews >>
- Last Updated: Jul 8, 2024 3:13 PM
- URL: https://guides.auraria.edu/researchmethods
1100 Lawrence Street Denver, CO 80204 303-315-7700 Ask Us Directions
Research Methods and Design
- Action Research
- Case Study Design
Literature Review
- Quantitative Research Methods
- Qualitative Research Methods
- Mixed Methods Study
- Indigenous Research and Ethics This link opens in a new window
- Identifying Empirical Research Articles This link opens in a new window
- Research Ethics and Quality
- Data Literacy
- Get Help with Writing Assignments
A literature review is a discussion of the literature (aka. the "research" or "scholarship") surrounding a certain topic. A good literature review doesn't simply summarize the existing material, but provides thoughtful synthesis and analysis. The purpose of a literature review is to orient your own work within an existing body of knowledge. A literature review may be written as a standalone piece or be included in a larger body of work.
You can read more about literature reviews, what they entail, and how to write one, using the resources below.
Am I the only one struggling to write a literature review?
Dr. Zina O'Leary explains the misconceptions and struggles students often have with writing a literature review. She also provides step-by-step guidance on writing a persuasive literature review.
An Introduction to Literature Reviews
Dr. Eric Jensen, Professor of Sociology at the University of Warwick, and Dr. Charles Laurie, Director of Research at Verisk Maplecroft, explain how to write a literature review, and why researchers need to do so. Literature reviews can be stand-alone research or part of a larger project. They communicate the state of academic knowledge on a given topic, specifically detailing what is still unknown.
This is the first video in a whole series about literature reviews. You can find the rest of the series in our SAGE database, Research Methods:
Videos covering research methods and statistics
Identify Themes and Gaps in Literature (with real examples) | Scribbr
Finding connections between sources is key to organizing the arguments and structure of a good literature review. In this video, you'll learn how to identify themes, debates, and gaps between sources, using examples from real papers.
4 Tips for Writing a Literature Review's Intro, Body, and Conclusion | Scribbr
While each review will be unique in its structure--based on both the existing body of both literature and the overall goals of your own paper, dissertation, or research--this video from Scribbr does a good job simplifying the goals of writing a literature review for those who are new to the process. In this video, you’ll learn what to include in each section, as well as 4 tips for the main body illustrated with an example.

- Literature Review This chapter in SAGE's Encyclopedia of Research Design describes the types of literature reviews and scientific standards for conducting literature reviews.
- UNC Writing Center: Literature Reviews This handout from the Writing Center at UNC will explain what literature reviews are and offer insights into the form and construction of literature reviews in the humanities, social sciences, and sciences.
- Purdue OWL: Writing a Literature Review The overview of literature reviews comes from Purdue's Online Writing Lab. It explains the basic why, what, and how of writing a literature review.
Organizational Tools for Literature Reviews
One of the most daunting aspects of writing a literature review is organizing your research. There are a variety of strategies that you can use to help you in this task. We've highlighted just a few ways writers keep track of all that information! You can use a combination of these tools or come up with your own organizational process. The key is choosing something that works with your own learning style.
Citation Managers
Citation managers are great tools, in general, for organizing research, but can be especially helpful when writing a literature review. You can keep all of your research in one place, take notes, and organize your materials into different folders or categories. Read more about citations managers here:
- Manage Citations & Sources
Concept Mapping
Some writers use concept mapping (sometimes called flow or bubble charts or "mind maps") to help them visualize the ways in which the research they found connects.

There is no right or wrong way to make a concept map. There are a variety of online tools that can help you create a concept map or you can simply put pen to paper. To read more about concept mapping, take a look at the following help guides:
- Using Concept Maps From Williams College's guide, Literature Review: A Self-guided Tutorial
Synthesis Matrix
A synthesis matrix is is a chart you can use to help you organize your research into thematic categories. By organizing your research into a matrix, like the examples below, can help you visualize the ways in which your sources connect.
- Walden University Writing Center: Literature Review Matrix Find a variety of literature review matrix examples and templates from Walden University.
- Writing A Literature Review and Using a Synthesis Matrix An example synthesis matrix created by NC State University Writing and Speaking Tutorial Service Tutors. If you would like a copy of this synthesis matrix in a different format, like a Word document, please ask a librarian. CC-BY-SA 3.0
- << Previous: Case Study Design
- Next: Quantitative Research Methods >>
- Last Updated: May 7, 2024 9:51 AM
CityU Home - CityU Catalog


- University of Texas Libraries
Literature Reviews
Steps in the literature review process.
- What is a literature review?
- Define your research question
- Determine inclusion and exclusion criteria
- Choose databases and search
- Review Results
- Synthesize Results
- Analyze Results
- Librarian Support
- Artificial Intelligence (AI) Tools
- You may need to some exploratory searching of the literature to get a sense of scope, to determine whether you need to narrow or broaden your focus
- Identify databases that provide the most relevant sources, and identify relevant terms (controlled vocabularies) to add to your search strategy
- Finalize your research question
- Think about relevant dates, geographies (and languages), methods, and conflicting points of view
- Conduct searches in the published literature via the identified databases
- Check to see if this topic has been covered in other discipline's databases
- Examine the citations of on-point articles for keywords, authors, and previous research (via references) and cited reference searching.
- Save your search results in a citation management tool (such as Zotero, Mendeley or EndNote)
- De-duplicate your search results
- Make sure that you've found the seminal pieces -- they have been cited many times, and their work is considered foundational
- Check with your professor or a librarian to make sure your search has been comprehensive
- Evaluate the strengths and weaknesses of individual sources and evaluate for bias, methodologies, and thoroughness
- Group your results in to an organizational structure that will support why your research needs to be done, or that provides the answer to your research question
- Develop your conclusions
- Are there gaps in the literature?
- Where has significant research taken place, and who has done it?
- Is there consensus or debate on this topic?
- Which methodological approaches work best?
- For example: Background, Current Practices, Critics and Proponents, Where/How this study will fit in
- Organize your citations and focus on your research question and pertinent studies
- Compile your bibliography
Note: The first four steps are the best points at which to contact a librarian. Your librarian can help you determine the best databases to use for your topic, assess scope, and formulate a search strategy.
Videos Tutorials about Literature Reviews
This 4.5 minute video from Academic Education Materials has a Creative Commons License and a British narrator.
Recommended Reading

- Last Updated: Aug 13, 2024 1:52 PM
- URL: https://guides.lib.utexas.edu/literaturereviews

Harvey Cushing/John Hay Whitney Medical Library
- Collections
- Research Help
YSN Doctoral Programs: Steps in Conducting a Literature Review
- Biomedical Databases
- Global (Public Health) Databases
- Soc. Sci., History, and Law Databases
- Grey Literature
- Trials Registers
- Data and Statistics
- Public Policy
- Google Tips
- Recommended Books
- Steps in Conducting a Literature Review
What is a literature review?
A literature review is an integrated analysis -- not just a summary-- of scholarly writings and other relevant evidence related directly to your research question. That is, it represents a synthesis of the evidence that provides background information on your topic and shows a association between the evidence and your research question.
A literature review may be a stand alone work or the introduction to a larger research paper, depending on the assignment. Rely heavily on the guidelines your instructor has given you.
Why is it important?
A literature review is important because it:
- Explains the background of research on a topic.
- Demonstrates why a topic is significant to a subject area.
- Discovers relationships between research studies/ideas.
- Identifies major themes, concepts, and researchers on a topic.
- Identifies critical gaps and points of disagreement.
- Discusses further research questions that logically come out of the previous studies.
APA7 Style resources

APA Style Blog - for those harder to find answers
1. Choose a topic. Define your research question.
Your literature review should be guided by your central research question. The literature represents background and research developments related to a specific research question, interpreted and analyzed by you in a synthesized way.
- Make sure your research question is not too broad or too narrow. Is it manageable?
- Begin writing down terms that are related to your question. These will be useful for searches later.
- If you have the opportunity, discuss your topic with your professor and your class mates.
2. Decide on the scope of your review
How many studies do you need to look at? How comprehensive should it be? How many years should it cover?
- This may depend on your assignment. How many sources does the assignment require?
3. Select the databases you will use to conduct your searches.
Make a list of the databases you will search.
Where to find databases:
- use the tabs on this guide
- Find other databases in the Nursing Information Resources web page
- More on the Medical Library web page
- ... and more on the Yale University Library web page
4. Conduct your searches to find the evidence. Keep track of your searches.
- Use the key words in your question, as well as synonyms for those words, as terms in your search. Use the database tutorials for help.
- Save the searches in the databases. This saves time when you want to redo, or modify, the searches. It is also helpful to use as a guide is the searches are not finding any useful results.
- Review the abstracts of research studies carefully. This will save you time.
- Use the bibliographies and references of research studies you find to locate others.
- Check with your professor, or a subject expert in the field, if you are missing any key works in the field.
- Ask your librarian for help at any time.
- Use a citation manager, such as EndNote as the repository for your citations. See the EndNote tutorials for help.
Review the literature
Some questions to help you analyze the research:
- What was the research question of the study you are reviewing? What were the authors trying to discover?
- Was the research funded by a source that could influence the findings?
- What were the research methodologies? Analyze its literature review, the samples and variables used, the results, and the conclusions.
- Does the research seem to be complete? Could it have been conducted more soundly? What further questions does it raise?
- If there are conflicting studies, why do you think that is?
- How are the authors viewed in the field? Has this study been cited? If so, how has it been analyzed?
Tips:
- Review the abstracts carefully.
- Keep careful notes so that you may track your thought processes during the research process.
- Create a matrix of the studies for easy analysis, and synthesis, across all of the studies.
- << Previous: Recommended Books
- Last Updated: Jun 20, 2024 9:08 AM
- URL: https://guides.library.yale.edu/YSNDoctoral
- Resources Home 🏠
- Try SciSpace Copilot
- Search research papers
- Add Copilot Extension
- Try AI Detector
- Try Paraphraser
- Try Citation Generator
- April Papers
- June Papers
- July Papers

Types of Literature Review — A Guide for Researchers

Table of Contents
Researchers often face challenges when choosing the appropriate type of literature review for their study. Regardless of the type of research design and the topic of a research problem , they encounter numerous queries, including:
What is the right type of literature review my study demands?
- How do we gather the data?
- How to conduct one?
- How reliable are the review findings?
- How do we employ them in our research? And the list goes on.
If you’re also dealing with such a hefty questionnaire, this article is of help. Read through this piece of guide to get an exhaustive understanding of the different types of literature reviews and their step-by-step methodologies along with a dash of pros and cons discussed.
Heading from scratch!
What is a Literature Review?
A literature review provides a comprehensive overview of existing knowledge on a particular topic, which is quintessential to any research project. Researchers employ various literature reviews based on their research goals and methodologies. The review process involves assembling, critically evaluating, and synthesizing existing scientific publications relevant to the research question at hand. It serves multiple purposes, including identifying gaps in existing literature, providing theoretical background, and supporting the rationale for a research study.
What is the importance of a Literature review in research?
Literature review in research serves several key purposes, including:
- Background of the study: Provides proper context for the research. It helps researchers understand the historical development, theoretical perspectives, and key debates related to their research topic.
- Identification of research gaps: By reviewing existing literature, researchers can identify gaps or inconsistencies in knowledge, paving the way for new research questions and hypotheses relevant to their study.
- Theoretical framework development: Facilitates the development of theoretical frameworks by cultivating diverse perspectives and empirical findings. It helps researchers refine their conceptualizations and theoretical models.
- Methodological guidance: Offers methodological guidance by highlighting the documented research methods and techniques used in previous studies. It assists researchers in selecting appropriate research designs, data collection methods, and analytical tools.
- Quality assurance and upholding academic integrity: Conducting a thorough literature review demonstrates the rigor and scholarly integrity of the research. It ensures that researchers are aware of relevant studies and can accurately attribute ideas and findings to their original sources.
Types of Literature Review
Literature review plays a crucial role in guiding the research process , from providing the background of the study to research dissemination and contributing to the synthesis of the latest theoretical literature review findings in academia.
However, not all types of literature reviews are the same; they vary in terms of methodology, approach, and purpose. Let's have a look at the various types of literature reviews to gain a deeper understanding of their applications.
1. Narrative Literature Review
A narrative literature review, also known as a traditional literature review, involves analyzing and summarizing existing literature without adhering to a structured methodology. It typically provides a descriptive overview of key concepts, theories, and relevant findings of the research topic.
Unlike other types of literature reviews, narrative reviews reinforce a more traditional approach, emphasizing the interpretation and discussion of the research findings rather than strict adherence to methodological review criteria. It helps researchers explore diverse perspectives and insights based on the research topic and acts as preliminary work for further investigation.
Steps to Conduct a Narrative Literature Review

Source:- https://www.researchgate.net/figure/Steps-of-writing-a-narrative-review_fig1_354466408
Define the research question or topic:
The first step in conducting a narrative literature review is to clearly define the research question or topic of interest. Defining the scope and purpose of the review includes — What specific aspect of the topic do you want to explore? What are the main objectives of the research? Refine your research question based on the specific area you want to explore.
Conduct a thorough literature search
Once the research question is defined, you can conduct a comprehensive literature search. Explore and use relevant databases and search engines like SciSpace Discover to identify credible and pertinent, scholarly articles and publications.
Select relevant studies
Before choosing the right set of studies, it’s vital to determine inclusion (studies that should possess the required factors) and exclusion criteria for the literature and then carefully select papers. For example — Which studies or sources will be included based on relevance, quality, and publication date?
*Important (applies to all the reviews): Inclusion criteria are the factors a study must include (For example: Include only peer-reviewed articles published between 2022-2023, etc.). Exclusion criteria are the factors that wouldn’t be required for your search strategy (Example: exclude irrelevant papers, preprints, written in non-English, etc.)
Critically analyze the literature
Once the relevant studies are shortlisted, evaluate the methodology, findings, and limitations of each source and jot down key themes, patterns, and contradictions. You can use efficient AI tools to conduct a thorough literature review and analyze all the required information.
Synthesize and integrate the findings
Now, you can weave together the reviewed studies, underscoring significant findings such that new frameworks, contrasting viewpoints, and identifying knowledge gaps.
Discussion and conclusion
This is an important step before crafting a narrative review — summarize the main findings of the review and discuss their implications in the relevant field. For example — What are the practical implications for practitioners? What are the directions for future research for them?
Write a cohesive narrative review
Organize the review into coherent sections and structure your review logically, guiding the reader through the research landscape and offering valuable insights. Use clear and concise language to convey key points effectively.
Structure of Narrative Literature Review
A well-structured, narrative analysis or literature review typically includes the following components:
- Introduction: Provides an overview of the topic, objectives of the study, and rationale for the review.
- Background: Highlights relevant background information and establish the context for the review.
- Main Body: Indexes the literature into thematic sections or categories, discussing key findings, methodologies, and theoretical frameworks.
- Discussion: Analyze and synthesize the findings of the reviewed studies, stressing similarities, differences, and any gaps in the literature.
- Conclusion: Summarizes the main findings of the review, identifies implications for future research, and offers concluding remarks.
Pros and Cons of Narrative Literature Review
- Flexibility in methodology and doesn’t necessarily rely on structured methodologies
- Follows traditional approach and provides valuable and contextualized insights
- Suitable for exploring complex or interdisciplinary topics. For example — Climate change and human health, Cybersecurity and privacy in the digital age, and more
- Subjectivity in data selection and interpretation
- Potential for bias in the review process
- Lack of rigor compared to systematic reviews
Example of Well-Executed Narrative Literature Reviews
Paper title: Examining Moral Injury in Clinical Practice: A Narrative Literature Review

Source: SciSpace
While narrative reviews offer flexibility, academic integrity remains paramount. So, ensure proper citation of all sources and maintain a transparent and factual approach throughout your critical narrative review, itself.
2. Systematic Review
A systematic literature review is one of the comprehensive types of literature review that follows a structured approach to assembling, analyzing, and synthesizing existing research relevant to a particular topic or question. It involves clearly defined criteria for exploring and choosing studies, as well as rigorous methods for evaluating the quality of relevant studies.
It plays a prominent role in evidence-based practice and decision-making across various domains, including healthcare, social sciences, education, health sciences, and more. By systematically investigating available literature, researchers can identify gaps in knowledge, evaluate the strength of evidence, and report future research directions.
Steps to Conduct Systematic Reviews

Source:- https://www.researchgate.net/figure/Steps-of-Systematic-Literature-Review_fig1_321422320
Here are the key steps involved in conducting a systematic literature review
Formulate a clear and focused research question
Clearly define the research question or objective of the review. It helps to centralize the literature search strategy and determine inclusion criteria for relevant studies.
Develop a thorough literature search strategy
Design a comprehensive search strategy to identify relevant studies. It involves scrutinizing scientific databases and all relevant articles in journals. Plus, seek suggestions from domain experts and review reference lists of relevant review articles.
Screening and selecting studies
Employ predefined inclusion and exclusion criteria to systematically screen the identified studies. This screening process also typically involves multiple reviewers independently assessing the eligibility of each study.
Data extraction
Extract key information from selected studies using standardized forms or protocols. It includes study characteristics, methods, results, and conclusions.
Critical appraisal
Evaluate the methodological quality and potential biases of included studies. Various tools (BMC medical research methodology) and criteria can be implemented for critical evaluation depending on the study design and research quetions .
Data synthesis
Analyze and synthesize review findings from individual studies to draw encompassing conclusions or identify overarching patterns and explore heterogeneity among studies.
Interpretation and conclusion
Interpret the findings about the research question, considering the strengths and limitations of the research evidence. Draw conclusions and implications for further research.
The final step — Report writing
Craft a detailed report of the systematic literature review adhering to the established guidelines of PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses). This ensures transparency and reproducibility of the review process.
By following these steps, a systematic literature review aims to provide a comprehensive and unbiased summary of existing evidence, help make informed decisions, and advance knowledge in the respective domain or field.
Structure of a systematic literature review
A well-structured systematic literature review typically consists of the following sections:
- Introduction: Provides background information on the research topic, outlines the review objectives, and enunciates the scope of the study.
- Methodology: Describes the literature search strategy, selection criteria, data extraction process, and other methods used for data synthesis, extraction, or other data analysis..
- Results: Presents the review findings, including a summary of the incorporated studies and their key findings.
- Discussion: Interprets the findings in light of the review objectives, discusses their implications, and identifies limitations or promising areas for future research.
- Conclusion: Summarizes the main review findings and provides suggestions based on the evidence presented in depth meta analysis.
*Important (applies to all the reviews): Remember, the specific structure of your literature review may vary depending on your topic, research question, and intended audience. However, adhering to a clear and logical hierarchy ensures your review effectively analyses and synthesizes knowledge and contributes valuable insights for readers.
Pros and Cons of Systematic Literature Review
- Adopts rigorous and transparent methodology
- Minimizes bias and enhances the reliability of the study
- Provides evidence-based insights
- Time and resource-intensive
- High dependency on the quality of available literature (literature research strategy should be accurate)
- Potential for publication bias
Example of Well-Executed Systematic Literature Review
Paper title: Systematic Reviews: Understanding the Best Evidence For Clinical Decision-making in Health Care: Pros and Cons.

Read this detailed article on how to use AI tools to conduct a systematic review for your research!
3. Scoping Literature Review
A scoping literature review is a methodological review type of literature review that adopts an iterative approach to systematically map the existing literature on a particular topic or research area. It involves identifying, selecting, and synthesizing relevant papers to provide an overview of the size and scope of available evidence. Scoping reviews are broader in scope and include a diverse range of study designs and methodologies especially focused on health services research.
The main purpose of a scoping literature review is to examine the extent, range, and nature of existing studies on a topic, thereby identifying gaps in research, inconsistencies, and areas for further investigation. Additionally, scoping reviews can help researchers identify suitable methodologies and formulate clinical recommendations. They also act as the frameworks for future systematic reviews or primary research studies.
Scoping reviews are primarily focused on —
- Emerging or evolving topics — where the research landscape is still growing or budding. Example — Whole Systems Approaches to Diet and Healthy Weight: A Scoping Review of Reviews .
- Broad and complex topics : With a vast amount of existing literature.
- Scenarios where a systematic review is not feasible: Due to limited resources or time constraints.
Steps to Conduct a Scoping Literature Review
While Scoping reviews are not as rigorous as systematic reviews, however, they still follow a structured approach. Here are the steps:
Identify the research question: Define the broad topic you want to explore.
Identify Relevant Studies: Conduct a comprehensive search of relevant literature using appropriate databases, keywords, and search strategies.
Select studies to be included in the review: Based on the inclusion and exclusion criteria, determine the appropriate studies to be included in the review.
Data extraction and charting : Extract relevant information from selected studies, such as year, author, main results, study characteristics, key findings, and methodological approaches. However, it varies depending on the research question.
Collate, summarize, and report the results: Analyze and summarize the extracted data to identify key themes and trends. Then, present the findings of the scoping review in a clear and structured manner, following established guidelines and frameworks .
Structure of a Scoping Literature Review
A scoping literature review typically follows a structured format similar to a systematic review. It includes the following sections:
- Introduction: Introduce the research topic and objectives of the review, providing the historical context, and rationale for the study.
- Methods : Describe the methods used to conduct the review, including search strategies, study selection criteria, and data extraction procedures.
- Results: Present the findings of the review, including key themes, concepts, and patterns identified in the literature review.
- Discussion: Examine the implications of the findings, including strengths, limitations, and areas for further examination.
- Conclusion: Recapitulate the main findings of the review and their implications for future research, policy, or practice.
Pros and Cons of Scoping Literature Review
- Provides a comprehensive overview of existing literature
- Helps to identify gaps and areas for further research
- Suitable for exploring broad or complex research questions
- Doesn’t provide the depth of analysis offered by systematic reviews
- Subject to researcher bias in study selection and data extraction
- Requires careful consideration of literature search strategies and inclusion criteria to ensure comprehensiveness and validity.
In short, a scoping review helps map the literature on developing or emerging topics and identifying gaps. It might be considered as a step before conducting another type of review, such as a systematic review. Basically, acts as a precursor for other literature reviews.
Example of a Well-Executed Scoping Literature Review
Paper title: Health Chatbots in Africa Literature: A Scoping Review

Check out the key differences between Systematic and Scoping reviews — Evaluating literature review: systematic vs. scoping reviews
4. Integrative Literature Review
Integrative Literature Review (ILR) is a type of literature review that proposes a distinctive way to analyze and synthesize existing literature on a specific topic, providing a thorough understanding of research and identifying potential gaps for future research.
Unlike a systematic review, which emphasizes quantitative studies and follows strict inclusion criteria, an ILR embraces a more pliable approach. It works beyond simply summarizing findings — it critically analyzes, integrates, and interprets research from various methodologies (qualitative, quantitative, mixed methods) to provide a deeper understanding of the research landscape. ILRs provide a holistic and systematic overview of existing research, integrating findings from various methodologies. ILRs are ideal for exploring intricate research issues, examining manifold perspectives, and developing new research questions.
Steps to Conduct an Integrative Literature Review
- Identify the research question: Clearly define the research question or topic of interest as formulating a clear and focused research question is critical to leading the entire review process.
- Literature search strategy: Employ systematic search techniques to locate relevant literature across various databases and sources.
- Evaluate the quality of the included studies : Critically assess the methodology, rigor, and validity of each study by applying inclusion and exclusion criteria to filter and select studies aligned with the research objectives.
- Data Extraction: Extract relevant data from selected studies using a structured approach.
- Synthesize the findings : Thoroughly analyze the selected literature, identify key themes, and synthesize findings to derive noteworthy insights.
- Critical appraisal: Critically evaluate the quality and validity of qualitative research and included studies by using BMC medical research methodology.
- Interpret and present your findings: Discuss the purpose and implications of your analysis, spotlighting key insights and limitations. Organize and present the findings coherently and systematically.
Structure of an Integrative Literature Review
- Introduction : Provide an overview of the research topic and the purpose of the integrative review.
- Methods: Describe the opted literature search strategy, selection criteria, and data extraction process.
- Results: Present the synthesized findings, including key themes, patterns, and contradictions.
- Discussion: Interpret the findings about the research question, emphasizing implications for theory, practice, and prospective research.
- Conclusion: Summarize the main findings, limitations, and contributions of the integrative review.
Pros and Cons of Integrative Literature Review
- Informs evidence-based practice and policy to the relevant stakeholders of the research.
- Contributes to theory development and methodological advancement, especially in the healthcare arena.
- Integrates diverse perspectives and findings
- Time-consuming process due to the extensive literature search and synthesis
- Requires advanced analytical and critical thinking skills
- Potential for bias in study selection and interpretation
- The quality of included studies may vary, affecting the validity of the review
Example of Integrative Literature Reviews
Paper Title: An Integrative Literature Review: The Dual Impact of Technological Tools on Health and Technostress Among Older Workers

5. Rapid Literature Review
A Rapid Literature Review (RLR) is the fastest type of literature review which makes use of a streamlined approach for synthesizing literature summaries, offering a quicker and more focused alternative to traditional systematic reviews. Despite employing identical research methods, it often simplifies or omits specific steps to expedite the process. It allows researchers to gain valuable insights into current research trends and identify key findings within a shorter timeframe, often ranging from a few days to a few weeks — unlike traditional literature reviews, which may take months or even years to complete.
When to Consider a Rapid Literature Review?
- When time impediments demand a swift summary of existing research
- For emerging topics where the latest literature requires quick evaluation
- To report pilot studies or preliminary research before embarking on a comprehensive systematic review
Steps to Conduct a Rapid Literature Review
- Define the research question or topic of interest. A well-defined question guides the search process and helps researchers focus on relevant studies.
- Determine key databases and sources of relevant literature to ensure comprehensive coverage.
- Develop literature search strategies using appropriate keywords and filters to fetch a pool of potential scientific articles.
- Screen search results based on predefined inclusion and exclusion criteria.
- Extract and summarize relevant information from the above-preferred studies.
- Synthesize findings to identify key themes, patterns, or gaps in the literature.
- Prepare a concise report or a summary of the RLR findings.
Structure of a Rapid Literature Review
An effective structure of an RLR typically includes the following sections:
- Introduction: Briefly introduce the research topic and objectives of the RLR.
- Methodology: Describe the search strategy, inclusion and exclusion criteria, and data extraction process.
- Results: Present a summary of the findings, including key themes or patterns identified.
- Discussion: Interpret the findings, discuss implications, and highlight any limitations or areas for further research
- Conclusion: Summarize the key findings and their implications for practice or future research
Pros and Cons of Rapid Literature Review
- RLRs can be completed quickly, authorizing timely decision-making
- RLRs are a cost-effective approach since they require fewer resources compared to traditional literature reviews
- Offers great accessibility as RLRs provide prompt access to synthesized evidence for stakeholders
- RLRs are flexible as they can be easily adapted for various research contexts and objectives
- RLR reports are limited and restricted, not as in-depth as systematic reviews, and do not provide comprehensive coverage of the literature compared to traditional reviews.
- Susceptible to bias because of the expedited nature of RLRs. It would increase the chance of overlooking relevant studies or biases in the selection process.
- Due to time constraints, RLR findings might not be robust enough as compared to systematic reviews.
Example of a Well-Executed Rapid Literature Review
Paper Title: What Is the Impact of ChatGPT on Education? A Rapid Review of the Literature

A Summary of Literature Review Types
Literature Review Type | Narrative | Systematic | Integrative | Rapid | Scoping |
Approach | The traditional approach lacks a structured methodology | Systematic search, including structured methodology | Combines diverse methodologies for a comprehensive understanding | Quick review within time constraints | Preliminary study of existing literature |
How Exhaustive is the process? | May or may not be comprehensive | Exhaustive and comprehensive search | A comprehensive search for integration | Time-limited search | Determined by time or scope constraints |
Data Synthesis | Narrative | Narrative with tabular accompaniment | Integration of various sources or methodologies | Narrative and tabular | Narrative and tabular |
Purpose | Provides description of meta analysis and conceptualization of the review | Comprehensive evidence synthesis | Holistic understanding | Quick policy or practice guidelines review | Preliminary literature review |
Key characteristics | Storytelling, chronological presentation | Rigorous, traditional and systematic techniques approach | Diverse source or method integration | Time-constrained, systematic approach | Identifies literature size and scope |
Example Use Case | Historical exploration | Effectiveness evaluation | Quantitative, qualitative, and mixed combination | Policy summary | Research literature overview |
Tools and Resources for Conducting Different Types of Literature Reviews
Online scientific databases.
Platforms such as SciSpace , PubMed , Scopus , Elsevier , and Web of Science provide access to a vast array of scholarly literature, facilitating the search and data retrieval process.
Reference management software
Tools like SciSpace Citation Generator , EndNote, Zotero , and Mendeley assist researchers in organizing, annotating, and citing relevant literature, streamlining the review process altogether.
Automate Literature Review with AI tools
Automate the literature review process by using tools like SciSpace literature review which helps you compare and contrast multiple papers all on one screen in an easy-to-read matrix format. You can effortlessly analyze and interpret the review findings tailored to your study. It also supports the review in 75+ languages, making it more manageable even for non-English speakers.

Goes without saying — literature review plays a pivotal role in academic research to identify the current trends and provide insights to pave the way for future research endeavors. Different types of literature review has their own strengths and limitations, making them suitable for different research designs and contexts. Whether conducting a narrative review, systematic review, scoping review, integrative review, or rapid literature review, researchers must cautiously consider the objectives, resources, and the nature of the research topic.
If you’re currently working on a literature review and still adopting a manual and traditional approach, switch to the automated AI literature review workspace and transform your traditional literature review into a rapid one by extracting all the latest and relevant data for your research!
There you go!
Frequently Asked Questions
Narrative reviews give a general overview of a topic based on the author's knowledge. They may lack clear criteria and can be biased. On the other hand, systematic reviews aim to answer specific research questions by following strict methods. They're thorough but time-consuming.
A systematic review collects and analyzes existing research to provide an overview of a topic, while a meta-analysis statistically combines data from multiple studies to draw conclusions about the overall effect of an intervention or relationship between variables.
A systematic review thoroughly analyzes existing research on a specific topic using strict methods. In contrast, a scoping review offers a broader overview of the literature without evaluating individual studies in depth.
A systematic review thoroughly examines existing research using a rigorous process, while a rapid review provides a quicker summary of evidence, often by simplifying some of the systematic review steps to meet shorter timelines.
A systematic review carefully examines many studies on a single topic using specific guidelines. Conversely, an integrative review blends various types of research to provide a more comprehensive understanding of the topic.
You might also like

Boosting Citations: A Comparative Analysis of Graphical Abstract vs. Video Abstract

The Impact of Visual Abstracts on Boosting Citations

Introducing SciSpace’s Citation Booster To Increase Research Visibility
- Privacy Policy
Home » Literature Review – Types Writing Guide and Examples
Literature Review – Types Writing Guide and Examples
Table of Contents

Literature Review
Definition:
A literature review is a comprehensive and critical analysis of the existing literature on a particular topic or research question. It involves identifying, evaluating, and synthesizing relevant literature, including scholarly articles, books, and other sources, to provide a summary and critical assessment of what is known about the topic.
Types of Literature Review
Types of Literature Review are as follows:
- Narrative literature review : This type of review involves a comprehensive summary and critical analysis of the available literature on a particular topic or research question. It is often used as an introductory section of a research paper.
- Systematic literature review: This is a rigorous and structured review that follows a pre-defined protocol to identify, evaluate, and synthesize all relevant studies on a specific research question. It is often used in evidence-based practice and systematic reviews.
- Meta-analysis: This is a quantitative review that uses statistical methods to combine data from multiple studies to derive a summary effect size. It provides a more precise estimate of the overall effect than any individual study.
- Scoping review: This is a preliminary review that aims to map the existing literature on a broad topic area to identify research gaps and areas for further investigation.
- Critical literature review : This type of review evaluates the strengths and weaknesses of the existing literature on a particular topic or research question. It aims to provide a critical analysis of the literature and identify areas where further research is needed.
- Conceptual literature review: This review synthesizes and integrates theories and concepts from multiple sources to provide a new perspective on a particular topic. It aims to provide a theoretical framework for understanding a particular research question.
- Rapid literature review: This is a quick review that provides a snapshot of the current state of knowledge on a specific research question or topic. It is often used when time and resources are limited.
- Thematic literature review : This review identifies and analyzes common themes and patterns across a body of literature on a particular topic. It aims to provide a comprehensive overview of the literature and identify key themes and concepts.
- Realist literature review: This review is often used in social science research and aims to identify how and why certain interventions work in certain contexts. It takes into account the context and complexities of real-world situations.
- State-of-the-art literature review : This type of review provides an overview of the current state of knowledge in a particular field, highlighting the most recent and relevant research. It is often used in fields where knowledge is rapidly evolving, such as technology or medicine.
- Integrative literature review: This type of review synthesizes and integrates findings from multiple studies on a particular topic to identify patterns, themes, and gaps in the literature. It aims to provide a comprehensive understanding of the current state of knowledge on a particular topic.
- Umbrella literature review : This review is used to provide a broad overview of a large and diverse body of literature on a particular topic. It aims to identify common themes and patterns across different areas of research.
- Historical literature review: This type of review examines the historical development of research on a particular topic or research question. It aims to provide a historical context for understanding the current state of knowledge on a particular topic.
- Problem-oriented literature review : This review focuses on a specific problem or issue and examines the literature to identify potential solutions or interventions. It aims to provide practical recommendations for addressing a particular problem or issue.
- Mixed-methods literature review : This type of review combines quantitative and qualitative methods to synthesize and analyze the available literature on a particular topic. It aims to provide a more comprehensive understanding of the research question by combining different types of evidence.
Parts of Literature Review
Parts of a literature review are as follows:
Introduction
The introduction of a literature review typically provides background information on the research topic and why it is important. It outlines the objectives of the review, the research question or hypothesis, and the scope of the review.
Literature Search
This section outlines the search strategy and databases used to identify relevant literature. The search terms used, inclusion and exclusion criteria, and any limitations of the search are described.
Literature Analysis
The literature analysis is the main body of the literature review. This section summarizes and synthesizes the literature that is relevant to the research question or hypothesis. The review should be organized thematically, chronologically, or by methodology, depending on the research objectives.
Critical Evaluation
Critical evaluation involves assessing the quality and validity of the literature. This includes evaluating the reliability and validity of the studies reviewed, the methodology used, and the strength of the evidence.
The conclusion of the literature review should summarize the main findings, identify any gaps in the literature, and suggest areas for future research. It should also reiterate the importance of the research question or hypothesis and the contribution of the literature review to the overall research project.
The references list includes all the sources cited in the literature review, and follows a specific referencing style (e.g., APA, MLA, Harvard).
How to write Literature Review
Here are some steps to follow when writing a literature review:
- Define your research question or topic : Before starting your literature review, it is essential to define your research question or topic. This will help you identify relevant literature and determine the scope of your review.
- Conduct a comprehensive search: Use databases and search engines to find relevant literature. Look for peer-reviewed articles, books, and other academic sources that are relevant to your research question or topic.
- Evaluate the sources: Once you have found potential sources, evaluate them critically to determine their relevance, credibility, and quality. Look for recent publications, reputable authors, and reliable sources of data and evidence.
- Organize your sources: Group the sources by theme, method, or research question. This will help you identify similarities and differences among the literature, and provide a structure for your literature review.
- Analyze and synthesize the literature : Analyze each source in depth, identifying the key findings, methodologies, and conclusions. Then, synthesize the information from the sources, identifying patterns and themes in the literature.
- Write the literature review : Start with an introduction that provides an overview of the topic and the purpose of the literature review. Then, organize the literature according to your chosen structure, and analyze and synthesize the sources. Finally, provide a conclusion that summarizes the key findings of the literature review, identifies gaps in knowledge, and suggests areas for future research.
- Edit and proofread: Once you have written your literature review, edit and proofread it carefully to ensure that it is well-organized, clear, and concise.
Examples of Literature Review
Here’s an example of how a literature review can be conducted for a thesis on the topic of “ The Impact of Social Media on Teenagers’ Mental Health”:
- Start by identifying the key terms related to your research topic. In this case, the key terms are “social media,” “teenagers,” and “mental health.”
- Use academic databases like Google Scholar, JSTOR, or PubMed to search for relevant articles, books, and other publications. Use these keywords in your search to narrow down your results.
- Evaluate the sources you find to determine if they are relevant to your research question. You may want to consider the publication date, author’s credentials, and the journal or book publisher.
- Begin reading and taking notes on each source, paying attention to key findings, methodologies used, and any gaps in the research.
- Organize your findings into themes or categories. For example, you might categorize your sources into those that examine the impact of social media on self-esteem, those that explore the effects of cyberbullying, and those that investigate the relationship between social media use and depression.
- Synthesize your findings by summarizing the key themes and highlighting any gaps or inconsistencies in the research. Identify areas where further research is needed.
- Use your literature review to inform your research questions and hypotheses for your thesis.
For example, after conducting a literature review on the impact of social media on teenagers’ mental health, a thesis might look like this:
“Using a mixed-methods approach, this study aims to investigate the relationship between social media use and mental health outcomes in teenagers. Specifically, the study will examine the effects of cyberbullying, social comparison, and excessive social media use on self-esteem, anxiety, and depression. Through an analysis of survey data and qualitative interviews with teenagers, the study will provide insight into the complex relationship between social media use and mental health outcomes, and identify strategies for promoting positive mental health outcomes in young people.”
Reference: Smith, J., Jones, M., & Lee, S. (2019). The effects of social media use on adolescent mental health: A systematic review. Journal of Adolescent Health, 65(2), 154-165. doi:10.1016/j.jadohealth.2019.03.024
Reference Example: Author, A. A., Author, B. B., & Author, C. C. (Year). Title of article. Title of Journal, volume number(issue number), page range. doi:0000000/000000000000 or URL
Applications of Literature Review
some applications of literature review in different fields:
- Social Sciences: In social sciences, literature reviews are used to identify gaps in existing research, to develop research questions, and to provide a theoretical framework for research. Literature reviews are commonly used in fields such as sociology, psychology, anthropology, and political science.
- Natural Sciences: In natural sciences, literature reviews are used to summarize and evaluate the current state of knowledge in a particular field or subfield. Literature reviews can help researchers identify areas where more research is needed and provide insights into the latest developments in a particular field. Fields such as biology, chemistry, and physics commonly use literature reviews.
- Health Sciences: In health sciences, literature reviews are used to evaluate the effectiveness of treatments, identify best practices, and determine areas where more research is needed. Literature reviews are commonly used in fields such as medicine, nursing, and public health.
- Humanities: In humanities, literature reviews are used to identify gaps in existing knowledge, develop new interpretations of texts or cultural artifacts, and provide a theoretical framework for research. Literature reviews are commonly used in fields such as history, literary studies, and philosophy.
Role of Literature Review in Research
Here are some applications of literature review in research:
- Identifying Research Gaps : Literature review helps researchers identify gaps in existing research and literature related to their research question. This allows them to develop new research questions and hypotheses to fill those gaps.
- Developing Theoretical Framework: Literature review helps researchers develop a theoretical framework for their research. By analyzing and synthesizing existing literature, researchers can identify the key concepts, theories, and models that are relevant to their research.
- Selecting Research Methods : Literature review helps researchers select appropriate research methods and techniques based on previous research. It also helps researchers to identify potential biases or limitations of certain methods and techniques.
- Data Collection and Analysis: Literature review helps researchers in data collection and analysis by providing a foundation for the development of data collection instruments and methods. It also helps researchers to identify relevant data sources and identify potential data analysis techniques.
- Communicating Results: Literature review helps researchers to communicate their results effectively by providing a context for their research. It also helps to justify the significance of their findings in relation to existing research and literature.
Purpose of Literature Review
Some of the specific purposes of a literature review are as follows:
- To provide context: A literature review helps to provide context for your research by situating it within the broader body of literature on the topic.
- To identify gaps and inconsistencies: A literature review helps to identify areas where further research is needed or where there are inconsistencies in the existing literature.
- To synthesize information: A literature review helps to synthesize the information from multiple sources and present a coherent and comprehensive picture of the current state of knowledge on the topic.
- To identify key concepts and theories : A literature review helps to identify key concepts and theories that are relevant to your research question and provide a theoretical framework for your study.
- To inform research design: A literature review can inform the design of your research study by identifying appropriate research methods, data sources, and research questions.
Characteristics of Literature Review
Some Characteristics of Literature Review are as follows:
- Identifying gaps in knowledge: A literature review helps to identify gaps in the existing knowledge and research on a specific topic or research question. By analyzing and synthesizing the literature, you can identify areas where further research is needed and where new insights can be gained.
- Establishing the significance of your research: A literature review helps to establish the significance of your own research by placing it in the context of existing research. By demonstrating the relevance of your research to the existing literature, you can establish its importance and value.
- Informing research design and methodology : A literature review helps to inform research design and methodology by identifying the most appropriate research methods, techniques, and instruments. By reviewing the literature, you can identify the strengths and limitations of different research methods and techniques, and select the most appropriate ones for your own research.
- Supporting arguments and claims: A literature review provides evidence to support arguments and claims made in academic writing. By citing and analyzing the literature, you can provide a solid foundation for your own arguments and claims.
- I dentifying potential collaborators and mentors: A literature review can help identify potential collaborators and mentors by identifying researchers and practitioners who are working on related topics or using similar methods. By building relationships with these individuals, you can gain valuable insights and support for your own research and practice.
- Keeping up-to-date with the latest research : A literature review helps to keep you up-to-date with the latest research on a specific topic or research question. By regularly reviewing the literature, you can stay informed about the latest findings and developments in your field.
Advantages of Literature Review
There are several advantages to conducting a literature review as part of a research project, including:
- Establishing the significance of the research : A literature review helps to establish the significance of the research by demonstrating the gap or problem in the existing literature that the study aims to address.
- Identifying key concepts and theories: A literature review can help to identify key concepts and theories that are relevant to the research question, and provide a theoretical framework for the study.
- Supporting the research methodology : A literature review can inform the research methodology by identifying appropriate research methods, data sources, and research questions.
- Providing a comprehensive overview of the literature : A literature review provides a comprehensive overview of the current state of knowledge on a topic, allowing the researcher to identify key themes, debates, and areas of agreement or disagreement.
- Identifying potential research questions: A literature review can help to identify potential research questions and areas for further investigation.
- Avoiding duplication of research: A literature review can help to avoid duplication of research by identifying what has already been done on a topic, and what remains to be done.
- Enhancing the credibility of the research : A literature review helps to enhance the credibility of the research by demonstrating the researcher’s knowledge of the existing literature and their ability to situate their research within a broader context.
Limitations of Literature Review
Limitations of Literature Review are as follows:
- Limited scope : Literature reviews can only cover the existing literature on a particular topic, which may be limited in scope or depth.
- Publication bias : Literature reviews may be influenced by publication bias, which occurs when researchers are more likely to publish positive results than negative ones. This can lead to an incomplete or biased picture of the literature.
- Quality of sources : The quality of the literature reviewed can vary widely, and not all sources may be reliable or valid.
- Time-limited: Literature reviews can become quickly outdated as new research is published, making it difficult to keep up with the latest developments in a field.
- Subjective interpretation : Literature reviews can be subjective, and the interpretation of the findings can vary depending on the researcher’s perspective or bias.
- Lack of original data : Literature reviews do not generate new data, but rather rely on the analysis of existing studies.
- Risk of plagiarism: It is important to ensure that literature reviews do not inadvertently contain plagiarism, which can occur when researchers use the work of others without proper attribution.
About the author

Muhammad Hassan
Researcher, Academic Writer, Web developer
You may also like

Limitations in Research – Types, Examples and...

Research Process – Steps, Examples and Tips

Research Objectives – Types, Examples and...

Ethical Considerations – Types, Examples and...

Research Techniques – Methods, Types and Examples

Institutional Review Board – Application Sample...
- Locations and Hours
- UCLA Library
- Research Guides
- Biomedical Library Guides
Systematic Reviews
- Types of Literature Reviews
What Makes a Systematic Review Different from Other Types of Reviews?
- Planning Your Systematic Review
- Database Searching
- Creating the Search
- Search Filters and Hedges
- Grey Literature
- Managing and Appraising Results
- Further Resources
Reproduced from Grant, M. J. and Booth, A. (2009), A typology of reviews: an analysis of 14 review types and associated methodologies. Health Information & Libraries Journal, 26: 91–108. doi:10.1111/j.1471-1842.2009.00848.x
| Aims to demonstrate writer has extensively researched literature and critically evaluated its quality. Goes beyond mere description to include degree of analysis and conceptual innovation. Typically results in hypothesis or mode | Seeks to identify most significant items in the field | No formal quality assessment. Attempts to evaluate according to contribution | Typically narrative, perhaps conceptual or chronological | Significant component: seeks to identify conceptual contribution to embody existing or derive new theory | |
| Generic term: published materials that provide examination of recent or current literature. Can cover wide range of subjects at various levels of completeness and comprehensiveness. May include research findings | May or may not include comprehensive searching | May or may not include quality assessment | Typically narrative | Analysis may be chronological, conceptual, thematic, etc. | |
| Mapping review/ systematic map | Map out and categorize existing literature from which to commission further reviews and/or primary research by identifying gaps in research literature | Completeness of searching determined by time/scope constraints | No formal quality assessment | May be graphical and tabular | Characterizes quantity and quality of literature, perhaps by study design and other key features. May identify need for primary or secondary research |
| Technique that statistically combines the results of quantitative studies to provide a more precise effect of the results | Aims for exhaustive, comprehensive searching. May use funnel plot to assess completeness | Quality assessment may determine inclusion/ exclusion and/or sensitivity analyses | Graphical and tabular with narrative commentary | Numerical analysis of measures of effect assuming absence of heterogeneity | |
| Refers to any combination of methods where one significant component is a literature review (usually systematic). Within a review context it refers to a combination of review approaches for example combining quantitative with qualitative research or outcome with process studies | Requires either very sensitive search to retrieve all studies or separately conceived quantitative and qualitative strategies | Requires either a generic appraisal instrument or separate appraisal processes with corresponding checklists | Typically both components will be presented as narrative and in tables. May also employ graphical means of integrating quantitative and qualitative studies | Analysis may characterise both literatures and look for correlations between characteristics or use gap analysis to identify aspects absent in one literature but missing in the other | |
| Generic term: summary of the [medical] literature that attempts to survey the literature and describe its characteristics | May or may not include comprehensive searching (depends whether systematic overview or not) | May or may not include quality assessment (depends whether systematic overview or not) | Synthesis depends on whether systematic or not. Typically narrative but may include tabular features | Analysis may be chronological, conceptual, thematic, etc. | |
| Method for integrating or comparing the findings from qualitative studies. It looks for ‘themes’ or ‘constructs’ that lie in or across individual qualitative studies | May employ selective or purposive sampling | Quality assessment typically used to mediate messages not for inclusion/exclusion | Qualitative, narrative synthesis | Thematic analysis, may include conceptual models | |
| Assessment of what is already known about a policy or practice issue, by using systematic review methods to search and critically appraise existing research | Completeness of searching determined by time constraints | Time-limited formal quality assessment | Typically narrative and tabular | Quantities of literature and overall quality/direction of effect of literature | |
| Preliminary assessment of potential size and scope of available research literature. Aims to identify nature and extent of research evidence (usually including ongoing research) | Completeness of searching determined by time/scope constraints. May include research in progress | No formal quality assessment | Typically tabular with some narrative commentary | Characterizes quantity and quality of literature, perhaps by study design and other key features. Attempts to specify a viable review | |
| Tend to address more current matters in contrast to other combined retrospective and current approaches. May offer new perspectives | Aims for comprehensive searching of current literature | No formal quality assessment | Typically narrative, may have tabular accompaniment | Current state of knowledge and priorities for future investigation and research | |
| Seeks to systematically search for, appraise and synthesis research evidence, often adhering to guidelines on the conduct of a review | Aims for exhaustive, comprehensive searching | Quality assessment may determine inclusion/exclusion | Typically narrative with tabular accompaniment | What is known; recommendations for practice. What remains unknown; uncertainty around findings, recommendations for future research | |
| Combines strengths of critical review with a comprehensive search process. Typically addresses broad questions to produce ‘best evidence synthesis’ | Aims for exhaustive, comprehensive searching | May or may not include quality assessment | Minimal narrative, tabular summary of studies | What is known; recommendations for practice. Limitations | |
| Attempt to include elements of systematic review process while stopping short of systematic review. Typically conducted as postgraduate student assignment | May or may not include comprehensive searching | May or may not include quality assessment | Typically narrative with tabular accompaniment | What is known; uncertainty around findings; limitations of methodology | |
| Specifically refers to review compiling evidence from multiple reviews into one accessible and usable document. Focuses on broad condition or problem for which there are competing interventions and highlights reviews that address these interventions and their results | Identification of component reviews, but no search for primary studies | Quality assessment of studies within component reviews and/or of reviews themselves | Graphical and tabular with narrative commentary | What is known; recommendations for practice. What remains unknown; recommendations for future research |
- << Previous: Home
- Next: Planning Your Systematic Review >>
- Last Updated: Jul 23, 2024 3:40 PM
- URL: https://guides.library.ucla.edu/systematicreviews
Covidence website will be inaccessible as we upgrading our platform on Monday 23rd August at 10am AEST, / 2am CEST/1am BST (Sunday, 15th August 8pm EDT/5pm PDT)
How to write the methods section of a systematic review
Home | Blog | How To | How to write the methods section of a systematic review
Covidence breaks down how to write a methods section
The methods section of your systematic review describes what you did, how you did it, and why. Readers need this information to interpret the results and conclusions of the review. Often, a lot of information needs to be distilled into just a few paragraphs. This can be a challenging task, but good preparation and the right tools will help you to set off in the right direction 🗺️🧭.
Systematic reviews are so-called because they are conducted in a way that is rigorous and replicable. So it’s important that these methods are reported in a way that is thorough, clear, and easy to navigate for the reader – whether that’s a patient, a healthcare worker, or a researcher.
Like most things in a systematic review, the methods should be planned upfront and ideally described in detail in a project plan or protocol. Reviews of healthcare interventions follow the PRISMA guidelines for the minimum set of items to report in the methods section. But what else should be included? It’s a good idea to consider what readers will want to know about the review methods and whether the journal you’re planning to submit the work to has expectations on the reporting of methods. Finding out in advance will help you to plan what to include.
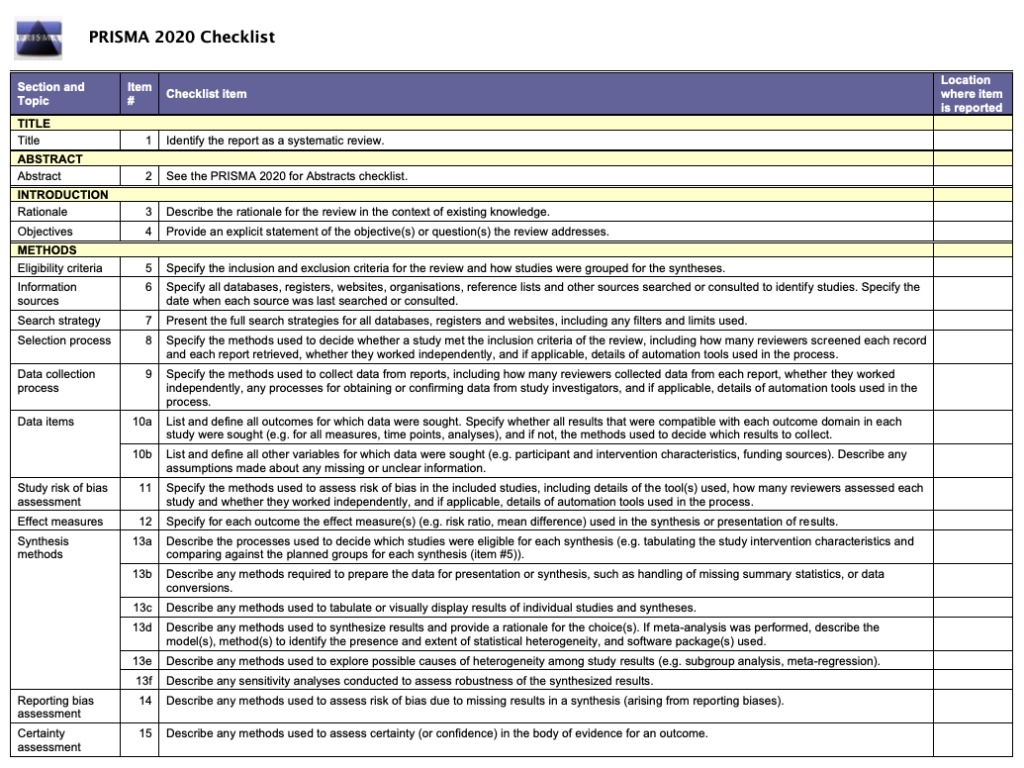
Describe what happened
While the research plan sets out what you intend to do, the methods section is a write-up of what actually happened. It’s not a simple case of rewriting the plan in the past tense – you will also need to discuss and justify deviations from the plan and describe the handling of issues that were unforeseen at the time the plan was written. For this reason, it is useful to make detailed notes before, during, and after the review is completed. Relying on memory alone risks losing valuable information and trawling through emails when the deadline is looming can be frustrating and time consuming!
Keep it brief
The methods section should be succinct but include all the noteworthy information. This can be a difficult balance to achieve. A useful strategy is to aim for a brief description that signposts the reader to a separate section or sections of supporting information. This could include datasets, a flowchart to show what happened to the excluded studies, a collection of search strategies, and tables containing detailed information about the studies.This separation keeps the review short and simple while enabling the reader to drill down to the detail as needed. And if the methods follow a well-known or standard process, it might suffice to say so and give a reference, rather than describe the process at length.
Follow a structure
A clear structure provides focus. Use of descriptive headings keeps the writing on track and helps the reader get to key information quickly. What should the structure of the methods section look like? As always, a lot depends on the type of review but it will certainly contain information relating to the following areas:
- Selection criteria ⭕
- Data collection and analysis 👩💻
- Study quality and risk of bias ⚖️
Let’s look at each of these in turn.
1. Selection criteria ⭕
The criteria for including and excluding studies are listed here. This includes detail about the types of studies, the types of participants, the types of interventions and the types of outcomes and how they were measured.
2. Search 🕵🏾♀️
Comprehensive reporting of the search is important because this means it can be evaluated and replicated. The search strategies are included in the review, along with details of the databases searched. It’s also important to list any restrictions on the search (for example, language), describe how resources other than electronic databases were searched (for example, non-indexed journals), and give the date that the searches were run. The PRISMA-S extension provides guidance on reporting literature searches.

Systematic reviewer pro-tip:
Copy and paste the search strategy to avoid introducing typos
3. Data collection and analysis 👩💻
This section describes:
- how studies were selected for inclusion in the review
- how study data were extracted from the study reports
- how study data were combined for analysis and synthesis
To describe how studies were selected for inclusion , review teams outline the screening process. Covidence uses reviewers’ decision data to automatically populate a PRISMA flow diagram for this purpose. Covidence can also calculate Cohen’s kappa to enable review teams to report the level of agreement among individual reviewers during screening.
To describe how study data were extracted from the study reports , reviewers outline the form that was used, any pilot-testing that was done, and the items that were extracted from the included studies. An important piece of information to include here is the process used to resolve conflict among the reviewers. Covidence’s data extraction tool saves reviewers’ comments and notes in the system as they work. This keeps the information in one place for easy retrieval ⚡.
To describe how study data were combined for analysis and synthesis, reviewers outline the type of synthesis (narrative or quantitative, for example), the methods for grouping data, the challenges that came up, and how these were dealt with. If the review includes a meta-analysis, it will detail how this was performed and how the treatment effects were measured.
4. Study quality and risk of bias ⚖️
Because the results of systematic reviews can be affected by many types of bias, reviewers make every effort to minimise it and to show the reader that the methods they used were appropriate. This section describes the methods used to assess study quality and an assessment of the risk of bias across a range of domains.
Steps to assess the risk of bias in studies include looking at how study participants were assigned to treatment groups and whether patients and/or study assessors were blinded to the treatment given. Reviewers also report their assessment of the risk of bias due to missing outcome data, whether that is due to participant drop-out or non-reporting of the outcomes by the study authors.
Covidence’s default template for assessing study quality is Cochrane’s risk of bias tool but it is also possible to start from scratch and build a tool with a set of custom domains if you prefer.
Careful planning, clear writing, and a structured approach are key to a good methods section. A methodologist will be able to refer review teams to examples of good methods reporting in the literature. Covidence helps reviewers to screen references, extract data and complete risk of bias tables quickly and efficiently. Sign up for a free trial today!

Laura Mellor. Portsmouth, UK
Perhaps you'd also like....

Top 5 Tips for High-Quality Systematic Review Data Extraction
Data extraction can be a complex step in the systematic review process. Here are 5 top tips from our experts to help prepare and achieve high quality data extraction.

How to get through study quality assessment Systematic Review
Find out 5 tops tips to conducting quality assessment and why it’s an important step in the systematic review process.

How to extract study data for your systematic review
Learn the basic process and some tips to build data extraction forms for your systematic review with Covidence.
Better systematic review management
Head office, working for an institution or organisation.
Find out why over 350 of the world’s leading institutions are seeing a surge in publications since using Covidence!
Request a consultation with one of our team members and start empowering your researchers:
By using our site you consent to our use of cookies to measure and improve our site’s performance. Please see our Privacy Policy for more information.
- Process: Literature Reviews
- Literature Review
- Managing Sources
Ask a Librarian

Does your assignment or publication require that you write a literature review? This guide is intended to help you understand what a literature is, why it is worth doing, and some quick tips composing one.
Understanding Literature Reviews
What is a literature review .
Typically, a literature review is a written discussion that examines publications about a particular subject area or topic. Depending on disciplines, publications, or authors a literature review may be:
A summary of sources An organized presentation of sources A synthesis or interpretation of sources An evaluative analysis of sources
A Literature Review may be part of a process or a product. It may be:
A part of your research process A part of your final research publication An independent publication
Why do a literature review?
The Literature Review will place your research in context. It will help you and your readers:
Locate patterns, relationships, connections, agreements, disagreements, & gaps in understanding Identify methodological and theoretical foundations Identify landmark and exemplary works Situate your voice in a broader conversation with other writers, thinkers, and scholars
The Literature Review will aid your research process. It will help you to:
Establish your knowledge Understand what has been said Define your questions Establish a relevant methodology Refine your voice Situate your voice in the conversation
What does a literature review look like?
The Literature Review structure and organization may include sections such as:
An introduction or overview A body or organizational sub-divisions A conclusion or an explanation of significance
The body of a literature review may be organized in several ways, including:
Chronologically: organized by date of publication Methodologically: organized by type of research method used Thematically: organized by concept, trend, or theme Ideologically: organized by belief, ideology, or school of thought

- Find a focus
- Find models
- Review your target publication
- Track citations
- Read critically
- Manage your citations
- Ask friends, faculty, and librarians
Additional Sources
- Reviewing the literature. Project Planner.
- Literature Review: By UNC Writing Center
- PhD on Track
- CU Graduate Students Thesis & Dissertation Guidance
- CU Honors Thesis Guidance
- Next: Managing Sources >>
- University of Colorado Boulder Libraries
- Research Guides
- Research Strategies
- Last Updated: Jun 20, 2024 3:23 PM
- URL: https://libguides.colorado.edu/strategies/litreview
- © Regents of the University of Colorado

Teaching and Research guides
Literature reviews.
- Introduction
- Plan your search
- Where to search
- Refine and update your search
- Finding grey literature
- Writing the review
- Referencing
Research methods overview
Finding literature on research methodologies, sage research methods online.
- Get material not at RMIT
- Further help
What are research methods?
Research methodology is the specific strategies, processes, or techniques utilised in the collection of information that is created and analysed.
The methodology section of a research paper, or thesis, enables the reader to critically evaluate the study’s validity and reliability by addressing how the data was collected or generated, and how it was analysed.
Types of research methods
There are three main types of research methods which use different designs for data collection.
(1) Qualitative research
Qualitative research gathers data about lived experiences, emotions or behaviours, and the meanings individuals attach to them. It assists in enabling researchers to gain a better understanding of complex concepts, social interactions or cultural phenomena. This type of research is useful in the exploration of how or why things have occurred, interpreting events and describing actions.
Examples of qualitative research designs include:
- focus groups
- observations
- document analysis
- oral history or life stories
(2) Quantitative research
Quantitative research gathers numerical data which can be ranked, measured or categorised through statistical analysis. It assists with uncovering patterns or relationships, and for making generalisations. This type of research is useful for finding out how many, how much, how often, or to what extent.
Examples of quantitative research designs include:
- surveys or questionnaires
- observation
- document screening
- experiments
(3) Mixed method research
Mixed Methods research integrates both Qualitative research and Quantitative research. It provides a holistic approach combining and analysing the statistical data with deeper contextualised insights. Using Mixed Methods also enables triangulation, or verification, of the data from two or more sources.
Sometimes in your literature review, you might need to discuss and evaluate relevant research methodologies in order to justify your own choice of research methodology.
When searching for literature on research methodologies it is important to search across a range of sources. No single information source will supply all that you need. Selecting appropriate sources will depend upon your research topic.
Developing a robust search strategy will help reduce irrelevant results. It is good practice to plan a strategy before you start to search.
Search tips
(1) free text keywords.
Free text searching is the use of natural language words to conduct your search. Use selective free text keywords such as: phenomenological, "lived experience", "grounded theory", "life experiences", "focus groups", interview, quantitative, survey, validity, variance, correlation and statistical.
To locate books on your desired methodology, try LibrarySearch . Remember to use refine options such as books, ebooks, subject, and publication date.
(2) Subject headings in Databases
Databases categorise their records using subject terms, or a controlled vocabulary (thesaurus). These subject headings may be useful to use, in addition to utilising free text keywords in a database search.
Subject headings will differ across databases, for example, the PubMed database uses 'Qualitative Research' whilst the CINHAL database uses 'Qualitative Studies.'
(3) Limiting search results
Databases enable sets of results to be limited or filtered by specific fields, look for options such as Publication Type, Article Type, etc. and apply them to your search.
(4) Browse the Library shelves
To find books on research methods browse the Library shelves at call number 001.42
- SAGE Research Methods Online SAGE Research Methods Online (SRMO) is a research tool supported by a newly devised taxonomy that links content and methods terms. It provides the most comprehensive picture available today of research methods (quantitative, qualitative and mixed methods) across the social and behavioural sciences.
SAGE Research Methods Overview (2:07 min) by SAGE Publishing ( YouTube )
- << Previous: Referencing
- Next: Get material not at RMIT >>

- Last Updated: Aug 12, 2024 8:44 AM
- URL: https://rmit.libguides.com/literature-review

Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
Part 1: Introduction to research
5. Writing your literature review
Chapter outline.
- Reading results (16 minute read)
- Synthesizing information (16 minute read)
- Writing a literature review (18 minute read)
Content warning: examples in this chapter contain references to domestic violence and details on types of abuse, drug use, poverty, mental health, sexual harassment and details on harassing behaviors, children’s mental health, LGBTQ+ oppression and suicide, obesity, anti-poverty stigma, and psychotic disorders.
5.1 Reading results
Learning objectives.
Learners will be able to…
- Describe how statistical significance and confidence intervals demonstrate which results are most important
- Differentiate between qualitative and quantitative results in an empirical journal article
If you recall from section 3.1 , empirical journal articles are those that report the results of quantitative or qualitative data analyzed by the author. They follow a set structure—introduction, methods, results, discussion/conclusions. This section is about reading the most challenging section: results.
Read beyond the abstract
At this point, I have read hundreds of literature reviews written by students. One of the challenges I have noted is that students will report the results as summarized in the abstract, rather than the detailed findings laid out in the results section of the article. This poses a problem when you are writing a literature review because you need to provide specific and clear facts that support your reading of the literature. The abstract may say something like: “we found that poverty is associated with mental health status.” For your literature review, you want the details, not the summary. In the results section of the article, you may find a sentence that states: “children living in households experiencing poverty are three times more likely to have a mental health diagnosis.” This more specific statistical information provides a stronger basis on which to build the arguments in your literature review.
Using the summarized results in an abstract is an understandable mistake to make. The results section often contains figures and tables that may be challenging to understand. Often, without having completed more advanced coursework on statistical or qualitative analysis, some of the terminology, symbols, or diagrams may be difficult to comprehend. This section is all about how to read and interpret the results of an empirical (quantitative or qualitative) journal article. Our discussion here will be basic, and in parts three and four of the textbook, you will learn more about how to interpret results from statistical tests and qualitative data analysis.
Remember, this section only addresses empirical articles. Non-empirical articles (e.g., theoretical articles, literature reviews) don’t have results. They cite the analysis of raw data completed by other authors, not the person writing the journal article who is merely summarizing others’ work.

Quantitative results
Quantitative articles often contain tables, and scanning them is a good way to begin reading the results. A table usually provides a quick, condensed summary of the report’s key findings. Tables are a concise way to report large amounts of data. Some tables present descriptive information about a researcher’s sample (often the first table in a results section). These tables will likely contain frequencies (N) and percentages (%). For example, if gender happened to be an important variable for the researcher’s analysis, a descriptive table would show how many and what percent of all study participants are of a particular gender. Frequencies or “how many” will probably be listed as N, while the percent symbol (%) might be used to indicate percentages.
In a table presenting a causal relationship, two sets of variables are represented. The independent variable , or cause, and the dependent variable , the effect. We’ll go into more detail on variables in Chapter 8 . Independent variable attributes are typically presented in the table’s columns, while dependent variable attributes are presented in rows. This allows the reader to scan a table’s rows to see how values on the dependent variable change as the independent variable values change. Tables displaying results of quantitative analysis will also likely include some information about which relationships are significant or not. We will discuss the details of significance and p-values later in this section.
Let’s look at a specific example: Table 5.1. It presents the causal relationship between gender and experiencing harassing behaviors at work. In this example, gender is the independent variable (the cause) and the harassing behaviors listed are the dependent variables (the effects). [1] Therefore, we place gender in the table’s columns and harassing behaviors in the table’s rows.
Reading across the table’s top row, we see that 2.9% of women in the sample reported experiencing subtle or obvious threats to their safety at work, while 4.7% of men in the sample reported the same. We can read across each of the rows of the table in this way. Reading across the bottom row, we see that 9.4% of women in the sample reported experiencing staring or invasion of their personal space at work while just 2.3% of men in the sample reported having the same experience. We’ll discuss p values later in this section.
| Subtle or obvious threats to your safety | 2.9% | 4.7% | 0.623 |
| Being hit, pushed, or grabbed | 2.2% | 4.7% | 0.480 |
| Comments or behaviors that demean your gender | 6.5% | 2.3% | 0.184 |
| Comments or behaviors that demean your age | 13.8% | 9.3% | 0.407 |
| Staring or invasion of your personal space | 9.4% | 2.3% | 0.039 |
| Note: Sample size was 138 for women and 43 for men. | |||
While you can certainly scan tables for key results, they are often difficult to understand without reading the text of the article. The article and table were meant to complement each other, and the text should provide information on how the authors interpret their findings. The table is not redundant with the text of the results section. Additionally, the first table in most results sections is a summary of the study’s sample, which provides more background information on the study than information about hypotheses and findings. It is also a good idea to look back at the methods section of the article as the data analysis plan the authors outline should walk you through the steps they took to analyze their data which will inform how they report them in the results section.
Statistical significance
The statistics reported in Table 5.1 represent what the researchers found in their sample. The purpose of statistical analysis is usually to generalize from a the small number of people in a study’s sample to a larger population of people. Thus, the researchers intend to make causal arguments about harassing behaviors at workplaces beyond those covered in the sample.
Generalizing is key to understanding statistical significance . According to Cassidy and colleagues, (2019) [2] 89% of research methods textbooks in psychology define statistical significance incorrectly. This includes an early draft of this textbook which defined statistical significance as “the likelihood that the relationships we observe could be caused by something other than chance.” If you have previously had a research methods class, this might sound familiar to you. It certainly did to me!
But statistical significance is less about “random chance” than more about the null hypothesis . Basically, at the beginning of a study a researcher develops a hypothesis about what they expect to find, usually that there is a statistical relationship between two or more variables . The null hypothesis is the opposite. It is the hypothesis that there is no relationship between the variables in a research study. Researchers then can hopefully reject the null hypothesis because they find a relationship between the variables.
For example, in Table 5.1 researchers were examining whether gender impacts harassment. Of course, researchers assumed that women were more likely to experience harassment than men. The null hypothesis, then, would be that gender has no impact on harassment. Once we conduct the study, our results will hopefully lead us to reject the null hypothesis because we find that gender impacts harassment. We would then generalize from our study’s sample to the larger population of people in the workplace.
Statistical significance is calculated using a p-value which is obtained by comparing the statistical results with a hypothetical set of results if the researchers re-ran their study a large number of times. Keeping with our example, imagine we re-ran our study with different men and women from different workplaces hundreds and hundred of times and we assume that the null hypothesis is true that gender has no impact on harassment. If results like ours come up pretty often when the null hypothesis is true, our results probably don’t mean much. “The smaller the p-value, the greater the statistical incompatibility with the null hypothesis” (Wasserstein & Lazar, 2016, p. 131). [3] Generally, researchers in the social sciences have used 0.05 as the value at which a result is significant (p is less than 0.05) or not significant (p is greater than 0.05). The p-value 0.05 refers to if 5% of those hypothetical results from re-running our study show the same or more extreme relationships when the null hypothesis is true. Researchers, however, may choose a stricter standard such as 0.01 in which only 1% of those hypothetical results are more extreme or a more lenient standard like 0.1 in which 10% of those hypothetical results are more extreme than what was found in the study.
Let’s look back at Table 5.1. Which one of the relationships between gender and harassing behaviors is statistically significant? It’s the last one in the table, “staring or invasion of personal space,” whose p-value is 0.039 (under the p<0.05 standard to establish statistical significance). Again, this indicates that if we re-ran our study over and over again and gender did not impact staring/invasion of space (i.e., the null hypothesis was true), only 3.9% of the time would we find similar or more extreme differences between men and women than what we observed in our study. Thus, we conclude that for staring or invasion of space only , there is a statistically significant relationship.
For contrast, let’s look at “being pushed, hit, or grabbed” and run through the same analysis to see if it is statistically significant. If we re-ran our study over and over again and the null hypothesis was true, 48% of the time (p=.48) we would find similar or more extreme differences between men and women. That means these results are not statistically significant.
This discussion should also highlight a point we discussed previously: that it is important to read the full results section, rather than simply relying on the summary in the abstract. If the abstract stated that most tests revealed no statistically significant relationships between gender and harassment, you would have missed the detail on which behaviors were and were not associated with gender. Read the full results section! And don’t be afraid to ask for help from a professor in understanding what you are reading, as results sections are often not written to be easily understood.
Statistical significance and p-values have been critiqued recently for a number of reasons, including that they are misused and misinterpreted (Wasserstein & Lazar, 2016) [4] , that researchers deliberately manipulate their analyses to have significant results (Head et al., 2015) [5] , and factor into the difficulty scientists have today in reproducing many of the results of previous social science studies (Peng, 2015). [6] For this reason, we share these principles, adapted from those put forth by the American Statistical Association, [7] for understanding and using p-values in social science:
- P-values provide evidence against a null hypothesis.
- P-values do not indicate whether the results were produced by random chance alone or if the researcher’s hypothesis is true, though both are common misconceptions.
- Statistical significance can be detected in minuscule differences that have very little effect on the real world.
- Nuance is needed to interpret scientific findings, as a conclusion does not become true or false when the p-value passes from p=0.051 to p=0.049.
- Real-world decision-making must use more than reported p-values. It’s easy to run analyses of large datasets and only report the significant findings.
- Greater confidence can be placed in studies that pre-register their hypotheses and share their data and methods openly with the public.
- “By itself, a p-value does not provide a good measure of evidence regarding a model or hypothesis. For example, a p-value near 0.05 taken by itself offers only weak evidence against the null hypothesis. Likewise, a relatively large p-value does not imply evidence in favor of the null hypothesis; many other hypotheses may be equally or more consistent with the observed data” (Wasserstein & Lazar, 2016, p. 132).
Confidence intervals
Because of the limitations of p-values, scientists can use other methods to determine whether their models of the world are true. One common approach is to use a confidence interval , or a range of values in which the true value is likely to be found. Confidence intervals are helpful because, as principal #5 above points out, p-values do not measure the size of an effect (Greenland et al., 2016). [8] Remember, something that has very little impact on the world can be statistically significant, and the values in a confidence interval would be helpful. In our example from Table 5.1, imagine our analysis produced a confidence interval that women are 1.2-3.4x more likely to experience “staring or invasion of personal space” than men. As with p-values, calculation for a confidence interval compares what was found in one study with a hypothetical set of results if we repeated the study over and over again. If we calculated 95% confidence intervals for all of the hypothetical set of hundreds and hundreds of studies, that would be our confidence interval.
Confidence intervals are pretty intuitive. As of this writing, my wife and are expecting our second child. The doctor told us our due date was December 11th. But the doctor also told us that December 11th was only their best estimate. They were actually 95% sure our baby might be born any time in the 30-day period between November 27th and December 25th. Confidence intervals are often listed with a percentage, like 90% or 95%, and a range of values, such as between November 27th and December 25th. You can read that as: “we are 95% sure your baby will be born between November 27th and December 25th because we’ve studied hundreds of thousands of fetuses and mothers, and we’re 95% sure your baby will be within these two dates.”
Notice that we’re hedging our bets here by using words like “best estimate.” When testing hypotheses, social scientists generally phrase their findings in a tentative way, talking about what results “indicate” or “support,” rather than making bold statements about what their results “prove.” Social scientists have humility because they understand the limitations of their knowledge. In a literature review, using a single study or fact to “prove” an argument right or wrong is often a signal to the person reading your literature review (usually your professor) that you may not have appreciated the limitations of that study or its place in the broader literature on the topic. Strong arguments in a literature review include multiple facts and ideas that span across multiple studies.
You can learn more about creating tables, reading tables, and tests of statistical significance in a class focused exclusively on statistical analysis. We provide links to many free and openly licensed resources on statistics in Chapter 16 . For now, we hope this brief introduction to reading tables will improve your confidence in reading and understanding the results sections in quantitative empirical articles.
Qualitative results
Quantitative articles will contain a lot of numbers and the results of statistical tests demonstrating associations between those numbers. Qualitative articles, on the other hand, will consist mostly of quotations from participants. For most qualitative articles, the authors want to put their results in the words of their participants, as they are the experts. Articles that lack quotations make it difficult to assess whether the researcher interpreted the data in a trustworthy, unbiased manner. These types of articles may also indicate how often particular themes or ideas came up in the data, potentially reflective of how important they were to participants.
Authors often organize qualitative results by themes and subthemes. For example, see this snippet from the results section in Bonanno and Veselak (2019) [9] discussion parents’ attitudes towards child mental health information sources.
Data analysis revealed four themes related to participants’ abilities to access mental health help and information for their children, and parents’ levels of trust in these sources. These themes are: others’ firsthand experiences family and friends with professional experience, protecting privacy, and uncertainty about schools as information sources. Trust emerged as an overarching and unifying concept for all of these themes. Others’ firsthand experiences. Several participants reported seeking information from other parents who had experienced mental health struggles similar to their own children. They often referenced friends or family members who had been or would be good sources of information due to their own personal experiences. The following quote from Adrienne demonstrates the importance of firsthand experience: [I would only feel comfortable sharing concerns or asking for advice] if I knew that they had been in the same situation. (Adrienne) Similarly, Michelle said: And I talked to a friend of mine who has kids who have IEPs in the district to see, kind of, how did she go about it. (Michelle) … Friends/family with professional experience . Several respondents referred to friends or family members who had professional experience with or knowledge of child mental health and suggested that these individuals would be good sources of information. For example, Hannah said: Well, what happened with me was I have an uncle who’s a psychiatrist. Sometimes if he’s up in (a city to the north), he’s retired, I can call him sometimes and get information. (Hannah) Michelle, who was in nursing school, echoed this sentiment: At this point, [if my child’s behavioral difficulties continued], I would probably call one of my [nursing] professors. That’s what I’ve done in the past when I’ve needed help with certain things…I have a professor who I would probably consider a friend who I would probably talk to first. She has a big adolescent practice. (Michelle) (p. 402-403)
The terms in bold above refer to the key themes (i.e., qualitative results) that were present in the data. Researchers will state the process by which they interpret each theme, providing a definition and usually some quotations from research participants. Researchers will also draw connections between themes, note consensus or conflict over themes, and situate the themes within the study context.
Qualitative results are specific to the time, place, and culture in which they arise, so you will have to use your best judgment to determine whether these results are relevant to your study. For example, students in my class at Radford University in Southwest Virginia may be studying rural populations. Would a study on group homes in a large urban city transfer well to group homes in a rural area?
Maybe. But even if you were using data from a qualitative study in another rural area, are all rural areas the same? How is the client population and sociocultural context in the article similar or different to the one in your study? Qualitative studies have tremendous depth, but researchers must be intentional about drawing conclusions about one context based on a study in another context.
Key Takeaways
- The results section of empirical articles are often the most difficult to understand.
- To understand a quantitative results section, look for results that were statistically significant and examine the confidence interval, if provided.
- To understand a qualitative results section, look for definitions of themes or codes and use the quotations provided to understand the participants’ perspective.
Select a quantitative empirical article related to your topic.
- Write down the results the authors identify as statistically significant in the results section.
- How do the authors interpret their results in the discussion section?
- Do the authors provide enough information in the introduction for you to understand their results?
Select a qualitative empirical article relevant to your topic.
- Write down the key themes the authors identify and how they were defined by the participants.
5.2 Organizing information
- Describe how to use summary tables to organize information from empirical articles
- Describe how to use topical outlines to organize information from the literature reviews of articles you read
- Create a concept map that visualizes the key concepts and relationships relevant to your working question
- Use what you learn in the literature search to revise your working question
This section will introduce you to three tools scholars use to organize and synthesize (i.e., weave together) information from multiple sources. First, we will discuss how to build a summary table containing information from empirical articles that are highly relevant—from literature review, to methods and results—to your entire research proposal. These are articles you will need to know the details of back-to-front because they are so highly related to your proposed study.
Second, we’ll discuss what to do with the other articles you’ve downloaded. As we’ve discussed previously, you’re not going to read most of the sources you download from start-to-finish. Instead, you’ll look at the author’s literature review, key ideas, and skim for any relevant passages for your project. As you do so, you should create a topical outline that organizes all relevant facts you might use in your literature that you’ve collected from the abstract, literature review, and conclusion of the articles you’ve found. Of course, it is important to note the original source of the information you are citing.
Finally, we will revisit concept mapping as a technique for visualizing the concepts in your study. Altogether, these techniques should help you create intermediary products—documents you are not likely to show to anyone or turn in for a grade—but that are vital steps to a final research proposal.
Organizing empirical articles using a summary table
Your research proposal is an empirical project. You will collect raw data and analyze it to answer your question. Over the next few weeks, identify about 10 articles that are empirically similar to the study you want to conduct. If you plan on conducting surveys of practitioners, it’s a good idea for you to read in detail other articles that have used similar methods (sampling, measures, data analysis) and asked similar questions to your proposal. A summary table can help you organize these Top 10 articles: empirical articles that are highly relevant to your proposal and working question.
Using the annotations in Section 4.2 as a guide, create a spreadsheet or Word table with your annotation categories as columns and each source as new row. For example, I was searching for articles on using a specific educational technique in the literature. I wanted to know whether other researchers found positive results, how big their samples were, and whether they were conducted at a single school or across multiple schools. I looked through each empirical article on the topic and filled in a summary table. At the end, I could do an easy visual analysis and state that most studies revealed no significant results and that there were few multi-site studies. These arguments were then included in my literature review. These tables are similar to those you will find in a systematic review article.
A basic summary table is provided in Figure 5.1. A more detailed example is available from Elaine Gregersen’s blog , and you can download an Excel template from Raul Pacheco-Vega’s blog . Remember, although “the means of summarizing can vary, the key at this point is to make sure you understand what you’ve found and how it relates to your topic and research question” (Bennard et al., 2014, para. 10). [10] As you revisit and revise your working question over the next few weeks, think about those sources that are so relevant you need to understand every detail about them.
A good summary table will also ensure that when you cite these articles in your literature review, you are able to provide the necessary detail and context for readers to properly understand the results. For example, one of the common errors I see in student literature reviews is using a small, exploratory study to represent the truth about a larger population. You will also notice important differences in how variables are measured or how people are sampled, for instance, and these details are often the source of a good critical review of the literature.

- Using your folder of article PDFs from you’ve downloaded in previous exercises, identify which articles are likely to be most relevant to your proposed study. This may change as you revise your working question and study design over the next few weeks. Create a list of 10 articles that are highly relevant to the extent that you will need to remember key details from each section of the article.
- Create a spreadsheet for your summary table and save it in your project folder on your hard drive. Using one of the templates linked in this chapter, fill in the columns of your spreadsheet. Enter the information from one of the articles you’ve read so far. As you finalize your research question over the next few weeks, fill in your summary table with the 5 most relevant empirical articles on your topic.
Synthesizing facts using a topical outline
If we’re only reading 10 articles in detail, what do we do with the others? Raul Pacheco-Vega recommends using the AIC approach : read the abstract, introduction, and conclusion (and the discussion section, in empirical articles). For non-empirical articles, it’s a little less clear but the first few pages and last few pages of an article usually contain the author’s reading of the relevant literature and their principal conclusions. You may also want to skim the first and last sentence of each paragraph. Only read paragraphs in which you are likely to find information relevant to your working question. Skimming like this gives you the general point of the article, though you should read in detail the most valuable resource of all—another author’s literature review.
It’s impossible to read all of the literature about your topic. You will read about 10 articles in detail. For a few dozen more (there is no magic number), you will read the abstract, introduction, and conclusion, skim the rest of the article, but ultimately never read everything. Make the most out of the articles you do read by extracting as many facts as possible from each. You are starting your research project without a lot of knowledge of the topic you want to study, and by using the literature reviews provided in academic journal articles, you can gain a lot of knowledge about a topic in a short period of time. This way, by reading only a small number of articles, you are also reading their citations and synthesis of dozens of other articles as well.
As you read an article in detail, we suggest copying any facts you find relevant in a separate word processing document. Another idea is to copy anything you’ve annotated as background information in Section 4.2 into an outline. Copying and pasting from PDF to Word can be difficult because PDFs are image files, not documents. To make that easier, use the HTML version of the article, convert the PDF to Word in Adobe Acrobat or another PDF reader, or use the “paste special” command to paste the content into Word without formatting. If it’s an old PDF, you may have to simply type out the information you need. It can be a messy job, but having all of your facts in one place is very helpful when drafting your literature review.
You should copy and paste any fact or argument you consider important. Some good examples include definitions of concepts, statistics about the size of the social problem, and empirical evidence about the key variables in the research question, among countless others. It’s a good idea to consult with your professor and the course syllabus to understand what they are looking for when reading your literature review. Facts for your literature review are principally found in the introduction, results, and discussion section of an empirical article or at any point in a non-empirical article. Copy and paste into your notes anything you may want to use in your literature review.
Importantly, you must make sure you note the original source of each bit of information you copy. Nothing is worse than needing to track down a source for fact you read who-knows-where. If you found a statistic that the author used in the introduction, it almost certainly came from another source that the author cited in a footnote or internal citation. You will want to check the original source to make sure the author represented the information correctly. Moreover, you may want to read the original study to learn more about your topic and discover other sources relevant to your inquiry.
Assuming you have pulled all of the facts out of multiple articles, it’s time to start thinking about how these pieces of information relate to each other. Start grouping each fact into categories and subcategories as shown in Table 5.2. For example, a statistic stating that single adults who are homeless are more likely to be male may fit into a category of gender and homelessness. For each topic or subtopic you identify during your critical analysis of each paper, determine what those papers have in common. Likewise, determine which differ. If there are contradictory findings, you may be able to identify methodological or theoretical differences that could account for these contradictions. For example, one study may sample only high-income earners or those living in a rural area. Determine what general conclusions you can report about the topic or subtopic, based on all of the information you’ve found.
Create a separate document containing a topical outline that combines your facts from each source and organizes them by topic or category. As you include more facts and more sources in your topical outline, you will begin to see how each fact fits into a category and how categories are related to one another. Keep in mind that your category names may change over time, as may their definitions. This is a natural reflection of the learning you are doing.
A complete topical outline is a long list of facts arranged by category. As you step back from the outline, you should assess which topic areas for which you have enough research support to allow you to draw strong conclusions. You should also assess which areas you need to do more research in before you can write a robust literature review. The topical outline should serve as a transitional document between the notes you write on each source and the literature review you submit to your professor. It is important to note that they contain plagiarized information that is copied and pasted directly from the primary sources. In this case, it is not problematic because these are just notes and are not meant to be turned in as your own ideas. For your final literature review, you must paraphrase these sources to avoid plagiarism. More importantly, you should keep your voice and ideas front-and-center in what you write as this is your analysis of the literature. Make strong claims and support them thoroughly using facts you found in the literature. We will pick up the task of writing your literature review in section 5.3.
- In your folder full of article PDFs, look for the most relevant review articles. If you don’t have any, try to look for some. If there are none in your topic area, you can also use other non-empirical articles or empirical articles with long literature reviews (in the introduction and discussion sections).
- Create a word processing document for your topical outline and save it in your project folder on your hard drive. Using a review article, start copying facts you identified as Background Information or Results into your topical outline. Try to organize each fact by topic or theme. Make sure to copy the internal citation for the original source of each fact. For articles that do not use internal citations, create one using the information in the footnotes and references. As you finalize your research question over the next few weeks, skim the literature reviews of the articles you download for key facts and copy them into your topical outline.
Putting the pieces together: Building a concept map
Developing a concept map or mind map around your topic can be helpful in figuring out how the facts fit together. We talked about concept mapping briefly in Chapter 2 , when we were first thinking about your topic and sketching out what you already know about it. Concept mapping during the literature review stage of a research project builds on this foundation of knowledge and aims to improve the “description of the breadth and depth of literature in a domain of inquiry. It also facilitates identification of the number and nature of studies underpinning mapped relationships among concepts, thus laying the groundwork for systematic research reviews and meta-analyses” (Lesley, Floyd, & Oermann, 2002, p. 229). [11] Its purpose, like other question refinement methods, is to help you organize, prioritize, and integrate material into a workable research area—one that is interesting, answerable, feasible, objective, scholarly, original, and clear.
Think about the topics you created in your topic outline. How do they relate to one another? Within each topic, how do facts relate to one another? As you write down what you have, think about what you already know. What other related concepts do you not yet have information about? What relationships do you need to investigate further? Building a conceptual map should help you understand what you already know, what you need to learn next, and how you can organize a literature review.
This technique is illustrated in this YouTube video about concept mapping . You may want to indicate which concepts and relationships you’ve already found in your review and which ones you think might be true but haven’t found evidence of yet. Once you get a sense of how your concepts are related and which relationships are important to you, it’s time to revise your working question.
- Create a concept map using a pencil and paper.
- Identify the key ideas inside the literature, how they relate to one another, and the facts you know about them.
- Reflect on those areas you need to learn more about prior to writing your literature review.
- As you finalize your research question over the next few weeks, update your concept map and think about how you might organize it into a written literature review.
- Refer to the topics and headings you use in your topical outline and think about what literature you have that helps you understand each concept and relationship between them in your concept map.
Revising your working question
You should be revisiting your working question throughout the literature review process. As you continue to learn more about your topic, your question will become more specific and clearly worded. This is normal, and there is no way to shorten this process. Keep revising your question in order to ensure it will contribute something new to the literature on your topic, is relevant to your target population, and is feasible for you to conduct as a student project.
For example, perhaps your initial idea or interest is how to prevent obesity. After an initial search of the relevant literature, you realize the topic of obesity is too broad to adequately cover in the time you have to do your project. You decide to narrow your focus to causes of childhood obesity. After reading some articles on childhood obesity, you further narrow your search to the influence of family risk factors on overweight children. A potential research question might then be, “What maternal factors are associated with toddler obesity in the United States?” You would then need to return to the literature to find more specific studies related to the variables in this question (e.g. maternal factors, toddler, obesity, toddler obesity).
Similarly, after an initial literature search for a broad topic such as school performance or grades, examples of a narrow research question might be:
- “To what extent does parental involvement in children’s education relate to school performance over the course of the early grades?”
- “Do parental involvement levels differ by family social, demographic, and contextual characteristics?”
- “What forms of parent involvement are most highly correlated with children’s outcomes? What factors might influence the extent of parental involvement?” (Early Childhood Longitudinal Program, 2011). [12]
In either case, your literature search, working question, and understanding of the topic are constantly changing as your knowledge of the topic deepens. A literature review is an iterative process, one that stops, starts, and loops back on itself multiple times before completion. As research is a practice behavior of social workers, you should apply the same type of critical reflection to your inquiry as you would to your clinical or macro practice.
There are many ways to approach synthesizing literature. We’ve reviewed the following: summary tables, topical outlines, and concept maps. Other examples you may encounter include annotated bibliographies and synthesis matrices. As you are learning how to conduct research, find a method that works for you. Reviewing the literature is a core component of evidence-based practice in social work. See the resources below if you need some additional help:
Literature Reviews: Using a Matrix to Organize Research / Saint Mary’s University of Minnesota
Literature Review: Synthesizing Multiple Sources / Indiana University
Writing a Literature Review and Using a Synthesis Matrix / Florida International University
Sample Literature Reviews Grid / Complied by Lindsay Roberts
Literature review preparation: Creating a summary table . (Includes transcript) / Laura Killam
- You won’t read every article all the way through. For most articles, reading the abstract, introduction, and conclusion are enough to determine its relevance. It’s expected that you skim or search for relevant sections of each article without reading the whole thing.
- For articles where everything seems relevant, use a summary table to keep track of details. These are particularly helpful with empirical articles.
- For articles with literature review content relevant to your topic, copy any relevant information into a topical outline, along with the original source of that information.
- Use a concept map to help you visualize the key concepts in your topic area and the relationships between them.
- Revise your working question regularly. As you do, you will likely need to revise your search queries and include new articles.
- Look back at the working question for your topic and consider any necessary revisions. It is important that questions become clearer and more specific over time. It is also common that your working question shift over time, sometimes drastically, as you explore new lines of inquiry in the literature. Return to your working question regularly and make sure it reflects the focus of your inquiry. You will continue to revise your working question until we formalize it into a research question at the end of Part 2 of this textbook.
5.3 Writing your literature review
- Describe the components of a literature review
- Begin to write your literature review
- Identify the purpose of a problem statement
- Apply the components of a formal argument to your topic
- Use elements of formal writing style, including signposting and transitions
- Recognize commons errors in literature reviews
Congratulations! By now, you should have discovered, retrieved, evaluated, synthesized, and organized the information you need for your literature review. It’s now time to turn that stack of articles, papers, and notes into a literature review–it’s time to start writing!
Writing about research is different than other types of writing. Research writing is not like a journal entry or opinion paper. The goal here is not to apply your research question to your life or growth as a practitioner. Research writing is about the provision and interpretation of facts. The tone should be objective and unbiased, and personal experiences and opinions are excluded. Particularly for students who are used to writing case notes, research writing can be a challenge. That’s why its important to normalize getting help! If your professor has not built in peer review, consider setting up a peer review group among your peers. You should also reach out to your academic advisor to see if there are writing services on your campus available to graduate students. No one should feel bad for needing help with something they haven’t done before, haven’t done in a while, or were never taught how to do.
If you’ve followed the steps in this chapter, you likely have an outline, summary table, and concept map from which you can begin the writing process. But what do you need to include in your literature review? We’ve mentioned it before, but to summarize, a literature review should:
- Introduce the topic and define its key terms.
- Establish the importance of the topic.
- Provide an overview of the important literature related to the concepts found in the research question.
- Identify gaps or controversies in the literature.
- Point out consistent findings across studies.
- Synthesize that which is known about a topic, rather than just provide a summary of the articles you read.
- Discuss possible implications and directions for future research.
Do you have enough facts and sources to accomplish these tasks? It’s a good time to consult your outlines and notes on each article you plan to include in your literature review. You may also want to consult with your professor on what is expected of you. If there is something you are missing, you may want to jump back to section 2.3 where we discussed how to search for literature. While you can always fill in material, there is the danger that you will start writing without really knowing what you are talking about or what you want to say. For example, if you don’t have a solid definition of your key concepts or a sense of how the literature has developed over time, it will be difficult to make coherent scholarly claims about your topic.
There is no magical point at which one is ready to write. As you consider whether you are ready, it may be useful to ask yourself these questions:
- How will my literature review be organized?
- What section headings will I be using?
- How do the various studies relate to each other?
- What contributions do they make to the field?
- Where are the gaps or limitations in existing research?
- And finally, but most importantly, how does my own research fit into what has already been done?
The problem statement
Scholarly works often begin with a problem statement, which serves two functions. First, it establishes why your topic is a social problem worth studying. Second, it pulls your reader into the literature review. Who would want to read about something unimportant?

A problem statement generally answers the following questions, though these are far from exhaustive:
- Why is this an important problem to study?
- How many people are affected by this problem?
- How does this problem impact other social issues relevant to social work?
- Why is your target population an important one to study?
A strong problem statement, like the rest of your literature review, should be filled with empirical results, theory, and arguments based on the extant literature. A research proposal differs significantly from other more reflective essays you’ve likely completed during your social work studies. If your topic were domestic violence in rural Appalachia, I’m sure you could come up with answers to the above questions without looking at a single source. However, the purpose of the literature review is not to test your intuition, personal experience, or empathy. Instead, research methods are about gaining specific and articulable knowledge to inform action. With a problem statement, you can take a “boring” topic like the color of rooms used in an inpatient psychiatric facility, transportation patterns in major cities, or the materials used to manufacture baby bottles, and help others see the topic as you see it—an important part of the social world that impacts social work practice.
The structure of a literature review
In general, the problem statement belongs at the beginning of the literature review. We usually advise students to spend no more than a paragraph or two for a problem statement. For the rest of your literature review, there is no set formula by which it needs to be organized. However, a literature review generally follows the format of any other essay—Introduction, Body, and Conclusion.
The introduction to the literature review contains a statement or statements about the overall topic. At a minimum, the introduction should define or identify the general topic, issue, or area of concern. You might consider presenting historical background, mentioning the results of a seminal study, and providing definitions of important terms. The introduction may also point to overall trends in what has been previously published on the topic or on conflicts in theory, methodology, evidence, conclusions, or gaps in research and scholarship. We also suggest putting in a few sentences that walk the reader through the rest of the literature review. Highlight your main arguments from the body of the literature review and preview your conclusion. An introduction should let the reader know what to expect from the rest of your review.
The body of your literature review is where you demonstrate your synthesis and analysis of the literature. Again, do not just summarize the literature. We would also caution against organizing your literature review by source—that is, one paragraph for source A, one paragraph for source B, etc. That structure will likely provide an adequate summary of the literature you’ve found, but it would give you almost no synthesis of the literature. That approach doesn’t tell your reader how to put those facts together, it doesn’t highlight points of agreement or contention, or how each study builds on the work of others. In short, it does not demonstrate critical thinking.
Organize your review by argument
Instead, use your outlines and notes as a guide what you have to say about the important topics you need to cover. Literature reviews are written from the perspective of an expert in that field. After an exhaustive literature review, you should feel as though you are able to make strong claims about what is true—so make them! There is no need to hide behind “I believe” or “I think.” Put your voice out in front, loud and proud! But make sure you have facts and sources that back up your claims.
I’ve used the term “ argument ” here in a specific way. An argument in writing means more than simply disagreeing with what someone else said, as this classic Monty Python sketch demonstrates. Toulman, Rieke, and Janik (1984) identify six elements of an argument:
- Claim: the thesis statement—what you are trying to prove
- Grounds: theoretical or empirical evidence that supports your claim
- Warrant: your reasoning (rule or principle) connecting the claim and its grounds
- Backing: further facts used to support or legitimize the warrant
- Qualifier: acknowledging that the argument may not be true for all cases
- Rebuttal: considering both sides (as cited in Burnette, 2012) [13]
Let’s walk through an example. If I were writing a literature review on a negative income tax, a policy in which people in poverty receive an unconditional cash stipend from the government each month equal to the federal poverty level, I would want to lay out the following:
- Claim: the negative income tax is superior to other forms of anti-poverty assistance.
- Grounds: data comparing negative income tax recipients to people receiving anti-poverty assistance in existing programs, theory supporting a negative income tax, data from evaluations of existing anti-poverty programs, etc.
- Warrant: cash-based programs like the negative income tax are superior to existing anti-poverty programs because they allow the recipient greater self-determination over how to spend their money.
- Backing: data demonstrating the beneficial effects of self-determination on people in poverty.
- Qualifier: the negative income tax does not provide taxpayers and voters with enough control to make sure people in poverty are not wasting financial assistance on frivolous items.
- Rebuttal: policy should be about empowering the oppressed, not protecting the taxpayer, and there are ways of addressing taxpayer spending concerns through policy design.
Like any effective argument, your literature review must have some kind of structure. For example, it might begin by describing a phenomenon in a general way along with several studies that provide some detail, then describing two or more competing theories of the phenomenon, and finally presenting a hypothesis to test one or more of the theories. Or, it might describe one phenomenon, then describe another that seems inconsistent with the first, then propose a theory that resolves the inconsistency, and finally present a hypothesis to test that theory. In applied research, it might describe a phenomenon or theory, then describe how that phenomenon or theory applies to some important real-world situation, and finally, may suggest a way to test whether it does, in fact, apply to that situation.
Use signposts
Another important issue is signposting . It may not be a term you are familiar with, but you are likely familiar with the concept. Signposting refers to the words used to identify the organization and structure of your literature review to your reader. The most basic form of signposting is using a topic sentence at the beginning of each paragraph. A topic sentence introduces the argument you plan to make in that paragraph. For example, you might start a paragraph stating, “There is strong disagreement in the literature as to whether psychedelic drugs cause people to develop psychotic disorders, or whether psychotic disorders cause people to use psychedelic drugs.” Within that paragraph, your reader would likely assume you will present evidence for both arguments. The concluding sentence of your paragraph should address the topic sentence, discussing how the facts and arguments from the paragraph you’ve written support a specific conclusion. To continue with our example, I might say, “There is likely a reciprocal effect in which both the use of psychedelic drugs worsens pre-psychotic symptoms and worsening psychosis increases the desire to use psychedelic drugs.”

Signposting also involves using headings and subheadings. Your literature review will use APA formatting, which means you need to follow their rules for bolding, capitalization, italicization, and indentation of headings. Headings help your reader understand the structure of your literature review. They can also help if the reader gets lost and needs to re-orient themselves within the document. We often tell our students to assume we know nothing (they don’t mind) and need to be shown exactly where they are addressing each part of the literature review. It’s like walking a small child around, telling them “First we’ll do this, then we’ll do that, and when we’re done, we’ll know this!”
Another way to use signposting is to open each paragraph with a sentence that links the topic of the paragraph with the one before it. Alternatively, one could end each paragraph with a sentence that links it with the next paragraph. For example, imagine we wanted to link a paragraph about barriers to accessing healthcare with one about the relationship between the patient and physician. We could use a transition sentence like this: “Even if patients overcome these barriers to accessing care, the physician-patient relationship can create new barriers to positive health outcomes.” A transition sentence like this builds a connection between two distinct topics. Transition sentences are also useful within paragraphs. They tell the reader how to consider one piece of information in light of previous information. Even simple transitional words like ‘however’ and ‘similarly’ can help demonstrate critical thinking and link each building block of your argument together.
Many beginning researchers have difficulty incorporating transitions into their writing. Let’s look at an example. Instead of beginning a sentence or paragraph by launching into a description of a study, such as “Williams (2004) found that…,” it is better to start by indicating something about why you are describing this particular study. Here are some simple examples:
- Another example of this phenomenon comes from the work of Williams (2004)…
- Williams (2004) offers one explanation of this phenomenon…
- An alternative perspective has been provided by Williams (2004)…
Now that we know to use signposts, the natural question is “What goes on the signposts?” First, it is important to start with an outline of the main points that you want to make, organized in the order you want to make them. The basic structure of your argument should then be apparent from the outline itself. Unfortunately, there is no formula we can give you that will work for everyone, but we can provide some general pointers on structuring your literature review.
The literature review tends to move from general to more specific ideas. You can build a review by identifying areas of consensus and areas of disagreement. You may choose to present historical studies—preferably seminal studies that are of significant importance—and close with the most recent research. Another approach is to start with the most distantly related facts and literature and then report on those most closely related to your research question. You could also compare and contrast valid approaches, features, characteristics, theories – that is, one approach, then a second approach, followed by a third approach.
Here are some additional tips for writing the body of your literature review:
- Start broad and then narrow down to more specific information.
- When appropriate, cite two or more sources for a single point, but avoid long strings of references for a single idea.
- Use quotes sparingly. Quotations for definitions are okay, but reserve quotes for when something is said so well you couldn’t possible phrase it differently. Never use quotes for statistics.
- Paraphrase when you need to relay the specific details within an article
- Include only the aspects of the study that are relevant to your literature review. Don’t insert extra facts about a study just to take up space.
- Avoid reflective, personal writing. It is traditional to avoid using first-person language (I, we, us, etc.).
- Avoid informal language like contractions, idioms, and rhetorical questions.
- Note any sections of your review that lack citations from the literature. Your arguments need to be based in empirical or theoretical facts. Do not approach this like a reflective journal entry.
- Point out consistent findings and emphasize stronger studies over weaker ones.
- Point out important strengths and weaknesses of research studies, as well as contradictions and inconsistent findings.
- Implications and suggestions for further research (where there are gaps in the current literature) should be specific.
The conclusion should summarize your literature review, discuss implications, and create a space for further research needed in this area. Your conclusion, like the rest of your literature review, should make a point. What are the important implications of your literature review? How do they inform the question you are trying to answer?
You should consult with your professor and the course syllabus about the final structure your literature review should take. Here is an example of one possible structure:
- Establish the importance of the topic
- Number and type of people affected
- Seriousness of the impact
- Physical, psychological, economic, social, or spiritual consequences of the problem
- Definitions of key terms
- Supporting evidence
- Common findings across studies, gaps in the literature
- Research question(s) and hypothesis(es)
Editing your literature review
Literature reviews are more than a summary of the publications you find on a topic. As you have seen in this brief introduction, literature reviews represent a very specific type of research, analysis, and writing. We will explore these topics further in upcoming chapters. As you begin your literature review, here are some common errors to avoid:
- Accepting a researcher’s finding as valid without evaluating methodology and data
- Ignoring contrary findings and alternative interpretations
- Using findings that are not clearly related to your own study or using findings that are too general
- Dedicating insufficient time to literature searching
- Reporting statistical results from a single study, rather than synthesizing the results of multiple studies to provide a comprehensive view of the literature on a topic
- Relying too heavily on secondary sources
- Overusing quotations
- Not justifying arguments using specific facts or theories from the literature
For your literature review, remember that your goal is to construct an argument for the importance of your research question. As you start editing your literature review, make sure it is balanced. Accurately report common findings, areas where studies contradict each other, new theories or perspectives, and how studies cause us to reaffirm or challenge our understanding of your topic.
It is acceptable to argue that the balance of the research supports the existence of a phenomenon or is consistent with a theory (and that is usually the best that researchers in social work can hope for), but it is not acceptable to ignore contradictory evidence. A large part of what makes a research question interesting is uncertainty about its answer (University of Minnesota, 2016). [14]
In addition to subjectivity and bias, writer’s block can obstruct the completion of your literature review. Often times, writer’s block can stem from confusing the creating and editing parts of the writing process. Many writers often start by simply trying to type out what they want to say, regardless of how good it is. Author Anne Lamott (1995) [15] terms these “shitty first drafts,” and we all write them. They are a natural and important part of the writing process.
Even if you have a detailed outline from which to work, the words are not going to fall into place perfectly the first time you start writing. You should consider turning off the editing and critiquing part of your brain for a while and allow your thoughts to flow. Don’t worry about putting a correctly formatted internal citation (as long as you know which source you used there) when you first write. Just get the information out. Only after you’ve reached a natural stopping point might you go back and edit your draft for grammar, APA style, organization, flow, and more. Divorcing the writing and editing process can go a long way to addressing writer’s block—as can picking a topic about which you have something to say!
As you are editing, keep in mind these questions adapted from Green (2012): [16]
- Content: Have I clearly stated the main idea or purpose of the paper? Is the thesis or focus clearly presented and appropriate for the reader?
- Organization: How well is it structured? Is the organization spelled out and easy to follow for the reader ?
- Flow: Is there a logical flow from section to section, paragraph to paragraph, sentence to sentence? Are there transitions between and within paragraphs that link ideas together?
- Development: Have I validated the main idea with supporting material? Are supporting data sufficient? Does the conclusion match the introduction?
- Form: Are there any APA style issues, redundancy, problematic wording and terminology (always know the definition of any word you use!), flawed sentence constructions and selection, spelling, and punctuation?
Social workers use the APA style guide to format and structure their literature reviews. Most students know APA style only as it relates to internal and external citations. If you are confused about them, consult this amazing APA style guide from the University of Texas-Arlington library. Your university’s library likely has resources they created to help you with APA style, and you can meet with a librarian or your professor to talk about formatting questions you have. Make sure you budget in a few hours at the end of each project to build a correctly formatted references page and check your internal citations.
Of course, APA style is about much more than knowing there is a period after “et al.” or citing the location a book was published. APA style is also about what the profession considers to be good writing. If you haven’t picked up an APA publication manual because you use citation generators, know that I did the same thing when I was in school. Purchasing the APA manual can help you with a common problem we hear about from students. Every professor (and every website about APA style) seems to have their own peculiar idea of “correct” APA style that you can, if needed, demonstrate is not accurate.
Here are some additional resources, if you would like more guidance on writing your literature review.
Doing a literature review / University of Leicester
Get lit: The literature review / Texas A&M Writing Centre
Guidebook for social work literature reviews / by Rebecca Mauldin and Matthew DeCarlo
- A literature review is not a book report. Do not organize it by article, with one paragraph for each source in your references. Instead, organize it based on the key ideas and arguments.
- The problem statement draws the reader into your topic by highlighting the importance of the topic to social work and to society overall.
- Signposting is an important component of academic writing that helps your reader follow the structure of your argument and of your literature review.
- Transitions demonstrate critical thinking and help guide your reader through your arguments.
- Editing and writing are separate processes.
- Consult with an APA style guide or a librarian to help you format your paper.
Look at your professor’s prompt for the literature review component of your research proposal (or if you don’t have one, use the example question provided in this section).
- Write 2-3 facts you would use to address each question or component in the prompt.
- Reflect on which questions you have a lot of information about and which you need to gather more information about in order to answer adequately.
Outline the structure of your literature review using your concept map from Section 5.2 as a guide.
- Identify the key arguments you will make and how they are related to each other.
- Reflect on topic sentences and concluding sentences you would use for each argument.
Media Attributions
- Numbers © Pop and Zebra is licensed under a CC0 (Creative Commons Zero) license
- summary table © Laura Frederiksen is licensed under a Public Domain license
- problem-2731501_1920 © Geralt is licensed under a CC0 (Creative Commons Zero) license
- sign-2080927_1920 © MariaMichelle is licensed under a CC0 (Creative Commons Zero) license
- It wouldn’t make any sense to say that people’s workplace experiences cause their gender, so in this example, the question of which is the independent variable and which are the dependent variables has a pretty obvious answer. ↵
- Cassidy, S. A., Dimova, R., Giguère, B., Spence, J. R., & Stanley, D. J. (2019). Failing grade: 89% of introduction-to-psychology textbooks that define or explain statistical significance do so incorrectly. Advances in Methods and Practices in Psychological Science , 2 (3), 233-239. ↵
- Wasserstein, R. L., & Lazar, N. A. (2016). The ASA statement on p-values: context, process, and purpose. The American Statistician, 70 , p. 129-133. ↵
- Head, M. L., Holman, L., Lanfear, R., Kahn, A. T., & Jennions, M. D. (2015). The extent and consequences of p-hacking in science. PLoS biology, 13 (3). ↵
- Peng, R. (2015), The reproducibility crisis in science: A statistical counterattack. Significance , 12 , 30–32. ↵
- Greenland, S., Senn, S. J., Rothman, K. J., Carlin, J. B., Poole, C., Goodman, S. N., & Altman, D. G. (2016). Statistical tests, P values, confidence intervals, and power: a guide to misinterpretations. European journal of epidemiology , 31 (4), 337-350. ↵
- Bonanno, R., & Veselak, K. (2019). A matter of trust: Parents attitudes towards child mental health information sources. Advances in Social Work , 19 (2), 397-415. ↵
- Bernnard, D., Bobish, G., Hecker, J., Holden, I., Hosier, A., Jacobson, T., Loney, T., & Bullis, D. (2014). Presenting: Sharing what you’ve learned. In Bobish, G., & Jacobson, T. (eds.) The information literacy users guide: An open online textbook . https://milnepublishing.geneseo.edu/the-information-literacy-users-guide-an-open-online-textbook/chapter/present-sharing-what-youve-learned/ ↵
- Leslie, M., Floyd, J., & Oermann, M. (2002). Use of MindMapper software for research domain mapping. Computers, informatics, nursing, 20(6), 229-235. ↵
- Early Childhood Longitudinal Program. (2011). Example research questions . https://nces.ed.gov/ecls/researchquestions2011.asp ↵
- Burnett, D. (2012). Inscribing knowledge: Writing research in social work. In W. Green & B. L. Simon (Eds.), The Columbia guide to social work writing (pp. 65-82). New York, NY: Columbia University Press. ↵
- University of Minnesota Libraries Publishing. (2016). This is a derivative of Research Methods in Psychology by a publisher who has requested that they and the original author not receive attribution, which was originally released and is used under CC BY-NC-SA. This work, unless otherwise expressly stated, is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License ↵
- Lamott, A. (1995). Bird by bird: Some instructions on writing and life . New York, NY: Penguin. ↵
- Green, W. Writing strategies for academic papers. In W. Green & B. L. Simon (Eds.), The Columbia guide to social work writing (pp. 25-47). New York, NY: Columbia University Press. ↵
report the results of a quantitative or qualitative data analysis conducted by the author
a quick, condensed summary of the report’s key findings arranged by row and column
causes a change in the dependent variable
a variable that depends on changes in the independent variable
(as in generalization) to make claims about a large population based on a smaller sample of people or items
"Assuming that the null hypothesis is true and the study is repeated an infinite number times by drawing random samples from the same populations(s), less than 5% of these results will be more extreme than the current result" (Cassidy et al., 2019, p. 233).
the assumption that no relationship exists between the variables in question
“a logical grouping of attributes that can be observed and measured and is expected to vary from person to person in a population” (Gillespie & Wagner, 2018, p. 9)
summarizes the incompatibility between a particular set of data and a proposed model for the data, usually the null hypothesis. The lower the p-value, the more inconsistent the data are with the null hypothesis, indicating that the relationship is statistically significant.
a range of values in which the true value is likely to be, to provide a more accurate description of their data
a statement about what you think is true backed up by evidence and critical thinking
the words used to identify the organization and structure of your literature review to your reader
what a researcher hopes to accomplish with their study
Graduate research methods in social work Copyright © 2021 by Matthew DeCarlo, Cory Cummings, Kate Agnelli is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License , except where otherwise noted.
Share This Book
Systematic literature review of gender equity and social inclusion in primary education for teachers in Tanzania: assessing status and future directions
- Open access
- Published: 13 August 2024
- Volume 3 , article number 122 , ( 2024 )
Cite this article
You have full access to this open access article

- Henry Nkya 1 &
- Isack Kibona 2
Gender equity and social inclusion (GESI) are crucial for creating inclusive and equitable educational environments in primary schools. This systematic literature review aimed to interpret and synthesize the findings of previous studies on GESI interventions and programs in primary schools in Tanzania, identified gaps in the knowledge, and provided recommendations for policy and practice. A systematic literature review search identified 22 relevant studies that met the inclusion criteria. The studies conducted between 2010 and 2021, and the sample sizes of participants were above 50. More than 50% of the studies were conducted in rural areas and used a quasi-experimental design. The interventions evaluation included teacher training, community engagement, and curriculum reform. The systematic literature review employed statistical methods to measure effect sizes and employed traditional univariate systematic literature review to synthesize the results. A table summarizing the literature that met the inclusion criteria was created to ensure transparency and clarity in the data coding process. The systematic literature review found a positive effect of GESI interventions on various outcomes, including improved academic performance, reduced gender-based violence, and increased social inclusion. However, variations in effect sizes and study designs across the studies were noted. Several gaps were identified, such as the lack of long-term follow-up and the need for more rigorous study designs. The implications of the findings for policy and practice in promoting GESI in primary schools in Tanzania were discussed, and recommendations for future research were provided. This systematic literature review highlighted the importance of addressing GESI in primary school education in Tanzania and underscored the critical role of teachers in promoting these values. It calls for targeted interventions, policy enhancements, and further research to bridge the gaps identified in the literature.
Explore related subjects
- Artificial Intelligence
Avoid common mistakes on your manuscript.
1 Introduction
GESI are critical components of education that ensure equitable access to education for all individuals, regardless of their gender, socioeconomic status, ethnicity, or other backgrounds. In Tanzania, GESI has become a significant concern, particularly in primary schools, where gender and social inequalities often lead to disparities in educational outcomes.
Research has shown that girls are more likely to face barriers in education than boys, including poverty, early marriages, and cultural bias that prioritize boys’ education over girls [ 28 ]. Furthermore, children from marginalized groups, such as children with disabilities, children from ethnic minority groups, and children from low-income families, often experience unequal access to quality education [ 38 ].
Addressing GESI issues in primary schools is crucial for ensuring that all children have access to quality education, which is essential for their personal development and future success. GESI initiatives can promote equity and inclusion in schools and create an environment where all children feel valued and supported [ 13 ].
Addressing gender stereotypes in teacher education programs can play a vital role in promoting GESI in primary schools [ 32 ]. Similarly, Okkolin et al. [ 27 ] suggest that interventions that address GESI can improve educational outcomes for girls and marginalized groups.
Overall, promoting GESI in primary schools is essential for creating a more equitable and inclusive education system that benefits all children [ 2 ]. It requires a concerted effort from policymakers, educators, parents, and communities to work together to create a learning environment that is supportive, respectful, and inclusive for all children.
1.1 Theoretical framework
This study is guided by the Social Justice Theory, which emphasizes the need for equitable treatment, opportunities, and outcomes for all individuals, particularly those from marginalized and disadvantaged backgrounds [ 10 ]. This framework is crucial in understanding the components of GESI and their impact on educational outcomes. The Social Justice Theory aligns with the goals of GESI by promoting fairness and the elimination of disparities in education [ 1 ].
1.1.1 Components of GESI
The key components of GESI in this study include [ 24 ]:
Gender equity: ensuring that girls and boys have equal access to education and opportunities.
Social inclusion: creating an inclusive environment where all students, regardless of their backgrounds, can participate and succeed.
Teacher training: educating teachers on gender-sensitive and inclusive teaching practices.
Community engagement: involving communities in promoting GESI.
Curricula reform: developing and implementing curricula that address GESI issues.
1.2 Justification for focusing on Tanzania
Tanzania provides a unique context for examining GESI due to its diverse population and the significant challenges it faces in achieving GESI in education [ 18 ]. Despite efforts to promote GESI, disparities persist, making it an important area of study to identify effective interventions and inform policy and practice.
1.3 Rationale for conducting a systematic literature review
A Systematic literature review is an essential tool for synthesizing research findings from different studies and summarizing the overall effect size of an intervention or variable of interest [ 34 ]. Conducting a systematic literature review on GESI in primary school education is critical for providing an overview of the existing research and identifying gaps that need to be addressed in future research. It also helps establish the overall effect of interventions aimed at promoting GESI in primary schools in Tanzania [ 9 ]. The results of the Systematic literature review can inform policies and practices aimed at promoting GESI in primary school education, thereby improving learning outcomes for all children, regardless of their gender, social, and economic backgrounds.
By addressing the GESI issues and synthesizing the existing literature, this systematic literature review aims to contribute to a more equitable and inclusive educational environment in Tanzania [ 22 ].
1.4 Research objectives
To identify the state of GESI in primary schools. This objective aims to provide a comprehensive understanding of how GESI issues manifest in primary schools, considering various social and educational contexts.
To number factors that contribute to gender GESI in primary schools. This shall allow informed decisions on the effort to contain the issues of GESI.
To synthesize the findings of previous studies on GESI in primary schools. This objective focuses on aggregating and interpreting the results of existing research to offer a clear and cohesive picture of what is known about GESI interventions and their effectiveness.
To identify gaps in the knowledge of GESI in primary schools. By evaluating the existing literature, this objective seeks to highlight areas where further research is needed, identifying shortcomings in study designs, populations, or intervention strategies.
To provide recommendations for improving GESI in primary schools. Based on the synthesis of previous studies and identified gaps, this objective aims to propose actionable strategies and policies to enhance GESI in primary education.
2 Methodology
Having set the study objectives, the search-strategy for the study involved conducting a comprehensive literature review of studies on GESI in primary schools. The search was conducted using electronic databases such as Google Scholar, JSTOR, and EBSCOhost. The search terms used were “gender equity,” “social inclusion,” “primary schools,” “Tanzania,” and “teachers.” Additionally, hand searching was conducted by reviewing the reference lists of identified studies to identify any relevant studies that may have been missed during the initial search.
Inclusion criteria:
The study must be conducted in primary schools.
The study must focus on gender equity and/or social inclusion in education.
The study must involve teachers as the primary participants or focus on the teacher’s role in promoting GESI.
The study must be published in English between 2010 and 2022.
Exclusion criteria:
Studies conducted outside Tanzania.
Studies not related to gender equity and/or social inclusion in education.
Studies not involving teachers or not focusing on the teacher’s role in promoting GESI.
Studies published before 2010 or after 2022.
The search process was conducted by two independent reviewers to ensure the accuracy and completeness of the search results. The reviewers screened the titles and abstracts of the identified studies for relevance and then reviewed the full text of potentially relevant studies. Any discrepancies between the reviewers were resolved through discussion and consensus. Reviewers made necessary steps to ensure a justified systematic review. Overall, the Authors reviewed 22 papers considered to have met the set criteria.
2.1 Choice of the effect size measure and analytical methods
The effect size measure used in this study was generated by statistical tools, making it suitable for systematics review that synthesize findings across multiple studies. For similar research questions, the study employed traditional univariate meta-analysis. This method was chosen because it is suitable for synthesizing the results of multiple studies that investigate similar research questions. Traditional univariate meta-analysis allows for the calculation of an overall effect size, providing a comprehensive summary of the impact of GESI interventions across different studies.
2.2 Choice of software
We used R software, specifically the ‘metafor’ package, for our analysis. This software was selected due to its robustness and versatility in conducting analytical procedures. The ‘metafor’ package supports a wide range of meta-analytic models and methods, making it a comprehensive tool for this type of analysis.
2.3 Coding of effect sizes
Table 1 summarizes the literature included that meets the inclusion criteria. This table includes information such as study design, sample size, effect sizes, and any other relevant variables. This step ensures transparency and clarity in the data coding process.
3 Results and analysis
The layout of the manuscript has been organized accordingly, so that headings and subheadings clearly demarcates each step of the systematic literature review process.
3.1 Status of GESI in primary schools in Tanzania
3.1.1 persistent gender disparities.
One of the major findings in this study was that gender disparities in primary education persist in Tanzania. This was evident in the lower enrollment and completion rates for girls in primary schools compared to boys [ 36 ]. Girls are less likely to attend school than boys, with enrollment rates lower for girls at both the primary and secondary levels. Additionally, girls are more likely to drop out of school due to various reasons, including early marriage, household responsibilities, and financial constraints [ 5 ]. These disparities highlight the ongoing challenges faced by girls in accessing and completing primary education.
3.1.2 Cultural and societal beliefs
Several studies have identified cultural and societal beliefs as a major factor contributing to gender disparities in primary education. In many Tanzanian communities, girls are expected to prioritize domestic responsibilities over their education, which can lead to low enrollment rates and high drop-out rates [ 39 ]. Furthermore, gender-based violence and sexual harassment are prevalent in schools, with girls facing discrimination and harassment from both male students and teachers [ 4 ]. These issues underscore the need for targeted interventions to create a safer and more supportive educational environment for girls.
Furthermore, Losioki and Mdee [ 12 ] found that gender stereotypes perpetuated in teacher education programs in Tanzania, which can affect the ability of teachers to create a gender-equitable and socially inclusive classroom environment. Teachers may unconsciously reinforce gender stereotypes in the classroom, leading to further marginalization of girls and other vulnerable groups.
3.1.3 Underrepresented minorities
In addition, limited access to education for children with disabilities or those from low-income families and marginalized communities can perpetuate social inequalities in primary schools [ 30 ]. These students often face significant barriers, including inadequate school facilities, lack of appropriate learning materials, and insufficient support services, which hinder their educational progress.
3.2 Strategies addressing the challenge
Despite these challenges, there have been government efforts to improve GESI in primary schools. The government of Tanzania has committed to providing equal access to education for all children, regardless of gender, ethnicity, or socio-economic status. The government has implemented policies such as free primary education and affirmative action programs to promote equal access to education for all children, regardless of gender or social status [ 15 , 26 ]. These initiatives aim to reduce financial barriers to education and encourage the enrollment and retention of girls and children from marginalized groups. This includes initiatives such as the Tanzania Education Sector Development Plan (ESDP) and the Primary Education Development Program (PEDP) [ 6 , 16 ]. These programs aim to address systemic barriers in education and promote inclusive practices in schools. The government also is open to collaborate with external forces like international interventions, community development agencies and NGO to work toward enhancing GESI. Some Strategies Addressing GESI Challenges. For instance, projects that focus on community engagement and parental involvement have shown positive impacts in changing attitudes towards girls’ education and promoting inclusive practices [ 17 ].
3.2.1 International and community-based programs
In recent years, there have been an increase in programs and initiatives aimed at promoting GESI in primary education. For example, the “Let Girls Learn” program, launched by the US government in partnership with the Tanzanian government, aimed to increase access to education for girls and reduce gender disparities in education [ 7 ]. Similarly, the Tanzania Gender Networking Programme (TGNP) has been working to promote GESI in education through community mobilization, advocacy, and capacity building [ 14 ].
3.2.2 Interventions with recorded impact
Previous studies identified several approaches that have been successful in improving GESI in primary schools. Among others, at least two are discussed. One such approach is the use of gender-responsive pedagogy, which involves incorporating gender-sensitive teaching practices and materials into the classroom [ 17 ]. This method helps create a more inclusive learning environment that acknowledges and addresses the different needs of boys and girls. Another effective intervention is the provision of sanitary pads and menstrual hygiene education to girls, which has been shown to improve school attendance and reduce drop-out rates [ 35 ]. By addressing menstrual hygiene needs, schools can help ensure that girls do not miss out on education due to a lack of resources or stigma associated with menstruation.
3.2.3 Intervention recommendations
GESI are essential components of a quality education system, and there is a need to address the persistent gender disparities in primary education. While cultural and societal beliefs continue to be major barriers, efforts to improve GESI through government policies and initiatives, as well as community-based programs, showed promise. The use of gender-responsive pedagogy and the provision of menstrual hygiene education and supplies were promising approaches that showed positive results [ 21 ]. However, more research and investment are needed to ensure that all children have access to primary education. Continued collaboration between the government, NGOs, and communities is essential to sustain and expand these efforts, ensuring that all students can benefit from a supportive and equitable educational environment [ 29 ].
Overall, there is still much work to be done to ensure GESI in primary schools [ 33 ]. It will require continued efforts and collaboration from the government, educators, and communities to address cultural and traditional beliefs, promote teacher education that challenges gender stereotypes, and provide equal access to education for all children. Policymakers must prioritize the allocation of resources to support GESI initiatives and ensure that schools are equipped to meet the diverse needs of all students [ 3 ].
By addressing these systemic issues, Tanzania can make significant strides towards achieving an inclusive and equitable education system that benefits all children, irrespective of their gender or socioeconomic background. Continued research and monitoring are essential to evaluate the effectiveness of existing interventions and identify new strategies to overcome persistent challenges in promoting GESI in primary education [ 31 ].
3.2.4 Gaps in the knowledge about GESI in primary schools
While the literature have provided valuable insights into the state of GESI in primary schools in Tanzania, several gaps in the knowledge still need to be addressed.
One major gap is the lack of research on the experiences of marginalized groups, including children with disabilities and those from low-income households. Studies have shown that these groups face significant barriers to accessing education and are often excluded from educational opportunities. For example, a study by Mwaijande [ 20 ] found that children with disabilities faced challenges such as lack of access to assistive devices and negative attitudes from teachers and other students. Similarly, research by Pak et al. [ 30 ] and Thomas and Rugambwa [ 36 ] revealed that children from poor families often struggle to pay school fees and may not have access to basic learning materials.
Another gap in the Tanzanian knowledge is the lack of research on the experiences of female teachers in primary schools. While studies have examined gender stereotypes and biases among teacher education programs, Thomas and Rugambwa [ 36 ] stressed that there is limited research on the experiences of female teachers in the classroom. Research on female teachers could shed light on the ways in which gender intersects with other forms of marginalization, such as age and socioeconomic status.
Furthermore, there is a need for more research on effective interventions and strategies for promoting GESI in primary schools. While some studies have evaluated the impact of interventions such as teacher training programs [ 19 , 25 ] , more rigorous evaluations of these interventions are needed to determine their effectiveness and sustainability.
Additionally, there is a lack of longitudinal studies that follow the long-term impact of GESI interventions. Many studies focus on short-term outcomes, but understanding the lasting effects of interventions is crucial for developing sustainable policies and practices.
In summary, while previous research has provided valuable insights into GESI in primary schools, several gaps in the knowledge need to be addressed. Future research should focus on the experiences of marginalized groups, including children with disabilities and those from low-income households, as well as female teachers. Additionally, the study showed more need for more rigorous evaluations of interventions and strategies aimed at promoting GESI in primary schools. Longitudinal studies that assess the long-term impact of these interventions would also be beneficial.
3.3 Patterns observed across the studies
As observed in the study, there were some patterns and trends identified across the studies. Firstly, there was a consistent finding that gender disparities persist in primary schools, particularly in terms of access to education and academic achievement. Despite efforts to promote GESI, girls and marginalized groups continue to face significant barriers that hinder their educational progress.
Secondly, there was a growing recognition of the importance of addressing GESI in primary education, as evidenced by the increasing number of interventions and programs aimed at promoting these values. This trend indicates a positive shift towards acknowledging and addressing GESI issues within the education system.
Thirdly, the systematic literature review revealed that the role of teachers is critical in promoting GESI in primary schools. Teacher training and support are essential for equipping educators with the skills and knowledge needed to foster an inclusive and equitable learning environment. Studies consistently highlighted the need for gender-sensitive pedagogy and teacher professional development programs.
Finally, there were some gaps in the current knowledge base, particularly with regard to the long-term impact of interventions and the effectiveness of different approaches to promoting GESI in primary education. While some interventions showed promising results, more research was needed to determine their sustainability and broader applicability.
By addressing these gaps and building on the patterns observed across studies, future research could contribute to a more comprehensive understanding of GESI in primary schools and inform the development of policies and practices to promote equity and inclusion for all students.
To sum up, analysis revealed that GESI interventions have a positive effect on various outcomes such as academic performance, reduced gender-based violence, and increased social inclusion. However, variations in effect sizes and study designs were observed across the studies. The studies included in the systematic literature review used various designs, such as randomized controlled trials (RCTs) and quasi-experimental designs, which contributed to the diversity in effect sizes.
4 Discussion
GESI is a critical components of a better-quality education system over otherwise. In Tanzania, primary education is the foundation for future academic and professional success [ 23 ], making it essential to ensure that all students, regardless of gender or social status, have access to an inclusive and equitable education. Previous studies explored the state of GESI in primary schools and identified areas for improvement.
The findings of the study highlighted the state of GESI in primary schools. The analysis of some 10 included studies revealed that significant disparities in access to education and academic performance among genders persist, with girls being more disadvantaged. Additionally, children from marginalized backgrounds, such as those from low-income families or those with disabilities, face substantial barriers to education.
To sum up, the study suggests a holistic approach involving teachers, schools, communities, and policymakers. Thus, multifaceted approach is necessary to create a more inclusive and equitable education system. Therefore, Recommendations include:
Providing comprehensive teacher training on gender-sensitive teaching methods.
Implementing community-based initiatives to address social and cultural barriers.
Developing policies and programs prioritizing marginalized students’ needs.
4.1 Implications of the study
Overall, the systematic literature review provided important insights into the state of GESI in primary schools. While progress has been made, significant challenges remain. Continued efforts and investments are necessary to promote a more equitable and inclusive education system. Future research should address the identified gaps and build on the promising interventions highlighted in this study. Based on the evidence synthesized, it is clear that targeted interventions are necessary to address the barriers that girls and other marginalized groups face in accessing and completing primary education. The study has the following recommendations on policy and practice and the areas for future research.
4.1.1 Addressing school issues related to GESI
Teacher training: policies should mandate comprehensive training for teachers on gender-sensitive teaching practices. Educators need to be equipped with the skills and knowledge to foster an inclusive classroom environment that supports both boys and girls. This includes understanding how to address and counteract gender stereotypes and biases.
Providing resources: schools should be equipped with resources to support girls’ education. This includes the provision of sanitary pads, access to clean and safe gender-segregated toilets, and gender-sensitive teaching materials. These resources are essential in reducing barriers to attendance and participation for girls.
Reviewing curricula: the school curriculum should be reviewed and revised to promote GESI. Curricula should reflect the diversity of Tanzanian society and challenge existing gender stereotypes. Including content that promotes GESI will help inculcate these values in students from a young age.
4.1.2 Addressing structural and socio-economic barriers
Financial support: there should be policies to provide financial support to families who cannot afford school fees. This can include scholarships, free school meals, and other financial incentives that alleviate the economic burden on families and keep girls in school.
Cultural norms and attitudes: interventions must focus on changing cultural norms and attitudes that limit girls’ access to education. Community engagement and awareness campaigns are crucial in shifting perceptions and promoting the value of girls’ education. Programs should aim to involve parents and community leaders in promoting gender equity.
Reducing gender-based violence: schools should implement strict policies against gender-based violence and harassment. Providing a safe and supportive environment is crucial for retaining girls in school. Support services for victims of violence and harassment should be readily available.
4.1.3 Promoting girls’ participation and leadership
Extracurricular activities: schools should create opportunities for girls to engage in extracurricular activities. Programs such as sports, arts, and clubs can enhance girls’ skills and confidence, providing a platform for them to express themselves and develop leadership qualities.
Leadership training: providing leadership training for girls to support their involvement in decision-making processes within schools and communities is essential. This training can empower girls to take active roles in their schools and communities, fostering a sense of agency and leadership.
4.1.4 Comprehensive and integrated approach
Involving multiple stakeholders: a comprehensive approach to promoting GESI should involve multiple stakeholders, including the government, civil society, and communities. Collaboration among these groups is essential for creating a supportive environment for GESI.
Evidence-based interventions: policies and practices should be guided by evidence-based interventions tailored to the specific needs and contexts of different regions and populations. Utilizing data and research to inform practices ensures that efforts are effective and impactful.
Monitoring and evaluation: continuous monitoring and evaluation of interventions are necessary to assess their effectiveness and make necessary adjustments. This helps in ensuring the sustainability and scalability of successful initiatives.
The study highlights the importance of a comprehensive and integrated approach to promoting GESI in primary schools. It underscores the need for targeted interventions, policy enhancements, and continued efforts to address the persistent barriers that girls and marginalized groups face. By implementing these recommendations, Tanzania can make significant strides towards achieving a more inclusive and equitable education system for all children.
4.2 Areas for future research
Future research and policy efforts should focus on sustaining and scaling successful interventions, ensuring that all children, regardless of gender or socio-economic background, have access to quality education. Future research should address these gaps:
Experiences of marginalized groups: more high-quality research is needed on the experiences of marginalized groups, including children with disabilities and those from low-income households.
Female teachers: investigate the experiences of female teachers in primary schools to understand how gender intersects with other forms of marginalization, such as age and socioeconomic status.
Effectiveness of interventions: conduct more rigorous evaluations of specific interventions and strategies for promoting GESI, including long-term impact studies.
Intersectionality: explore the intersectionality of factors such as gender, socioeconomic status, and ethnicity to provide a more comprehensive
5 Conclusion
GESI is crucial for improving access to education, ensuring equal opportunities, and promoting positive social outcomes. Teachers play a critical role in promoting these values and must receive appropriate training and support to create inclusive learning environments. Policymakers and education leaders must prioritize efforts to address GESI in primary schools, including investing in research to understand the factors contributing to gender and social equality and identifying effective strategies for promoting GESI.
The systematic literature review examined the state of GESI in primary schools and revealed significant challenges, particularly in terms of teacher training and the implementation of policies and programs. The review highlighted persistent gender disparities and the barriers faced by marginalized groups, such as children with disabilities and those from low-income families.
The findings suggest that targeted interventions are needed to address these barriers, recommended interventions include:
Increasing access to education: efforts to increase access to education for marginalized groups, such as scholarships and school feeding programs.
Policy development: implementing policies that address gender-based violence and discrimination.
Community engagement: involving multiple stakeholders, including government, civil society, and communities, in promoting GESI.
Develop and implement teacher training programs: focus on GESI principles, awareness of gender biases, strategies for promoting inclusivity, and the use of gender-sensitive teaching materials.
Develop and implement gender-sensitive curricula: address gender biases and stereotypes across all subject areas.
Strengthen policies and regulations: enforce policies that promote GESI in school governance, teacher recruitment, and student enrollment.
Increase participation of girls: provide incentives for girls to attend school, such as scholarships and school feeding programs, and improve school infrastructure.
The study provides crucial insights into the state of GESI in primary schools and underscores the need for coordinated and sustained efforts to address these challenges. By implementing the recommended strategies and involving all stakeholders, Tanzania can ensure that all children have access to quality primary education that promotes GESI.
Data availability
No datasets were generated or analysed during the current study.
Adipat S, Chotikapanich R. Sustainable development goal 4: an education goal to achieve equitable quality education. Acad J Interdiscip Stud. 2022;11(6):174–83.
Article Google Scholar
Cavicchioni V, Motivans A. Monitoring educational disparities in less developed countries. In: In pursuit of equity in education: using international indicators to compare equity policies. New York: Springer; 2001. p. 217–40.
Google Scholar
Clancy J, Barnett A, Cecelski E, Pachauri S, Dutta S, Oparaocha S, Kooijman A. Gender in the transition to sustainable energy for all: from evidence to inclusive policies. 2019.
Colclough C, Rose P, Tembon M. Gender inequalities in primary schooling: the roles of poverty and adverse cultural practice. Int J Educ Dev. 2000;20(1):5–27.
Esteves M. Gender equality in education: a challenge for policy makers. Int J Soc Sci. 2018;4(2):893–905.
Fenech M, Skattebol J. Supporting the inclusion of low-income families in early childhood education: an exploration of approaches through a social justice lens. Int J Incl Educ. 2021;25(9):1042–60.
Frank A. Understanding the “success” of an all girls’ boarding school in rural Tanzania: perspectives of graduates, teachers, and administrators, PhD thesis. The Florida State University; 2019.
Group WB. Malawi systematic country diagnostic: breaking the cycle of low growth and slow poverty reduction. Washington, DC: World Bank; 2018.
Book Google Scholar
Guthridge M, Kirkman M, Penovic T, Giummarra MJ. Promoting gender equality: a systematic review of interventions. Soc Justice Res. 2022;35(3):318–43.
Kaur B. Equity and social justice in teaching and teacher education. Teach Teach Educ. 2012;28(4):485–92.
Lokina RB, Nyoni J, Kahyarara G. Social policy, gender and labour in Tanzania. Dar es Salaam: Economic and Social Research Foundation (ESRF); 2016.
Losioki BE, Mdee HK. The contribution of the hidden curriculum to gender inequality in teaching and learning materials: experiences from Tanzania. Asian J Educ Train. 2023;9(2):54–8.
Lovell E. Gender equality, social inclusion and resilience in Malawi. In: Building resilience and adapting to climate change. 2021.
Makulilo AB, Bakari M. Building a transformative feminist movement for women empowerment in Tanzania: the role of the Tanzania gender networking programme (TGNP-Mtandao). Afr Rev. 2021;13(2):155–74.
Malelu AM. Institutional factors influencing career advancement of women faculty: a case of, PhD thesis. Kenyatta University; 2015.
Mashala YL. The impact of the implementation of free education policy on secondary education in Tanzania. Int J Acad Multidiscip Res. 2019;3(1):6–14.
Mhewa, M. M., Bhalalusesa, E. P., & Kafanabo, E. (2021). Secondary school teachers’ understanding of gender-responsive pedagogy in bridging inequalities of students’ learning in tanzania. Papers in Education and Development, 38(2).
Mohun R, Biswas S, Jacobson J, Sajjad F. Infrastructure: a game changer for women’s economic empowerment. Background paper. UN Secretary-General’s High-Level Panel on Women’s Economic Empowerment; 2016.
Mondal S, Joe W, Akhauri S, Sinha I, Thakur P, Kumar V, Kumar T, Pradhan N, Kumar A. Delivering PACE++ curriculum in community settings: impact of TARA intervention on gender attitudes and dietary practices among adolescent girls in Bihar, India. PLoS ONE. 2023;18(11): e0293941.
Mwaijande VT. Access to education and assistive devices for children with physical disabilities in Tanzania, Master’s thesis. Oslo and Akershus University College; 2014.
Mwakabenga RJ, Komba SC. Gender inequalities in pedagogical classroom practice: what influence do teachers make? J Educ Humanit Sci. 2021;10(3):66–82.
Nazneen S, Cole N. Literature review on socially inclusive budgeting. 2018.
Ndijuye LG, Mligo IR, Machumu MAM. Early childhood education in Tanzania: views and beliefs of stakeholders on its status and development. Global Educ Rev. 2020;7(3):22–39.
Nelly S. Gender equality and social inclusion (GESI) in village development. Legal Brief. 2021;10(2):245–52.
Nkya HE, Bimbiga I. Unlocking potential: the positive impact of in-service training on science and mathematics teachers teaching strategies. Res Humanit Soc Sci. 2023. https://doi.org/10.7176/RHSS/13-16-04 .
Nyoni WP, He C, Yusuph ML. Sustainable interventions in enhancing gender parity in senior leadership positions in higher education in Tanzania. J Educ Pract. 2017;8(13):44–54.
Okkolin M-A, Lehtomäki E, Bhalalusesa E. The successful education sector development in Tanzania—comment on gender balance and inclusive education. Gend Educ. 2010;22(1):63–71.
Omari CK, Mbilinyi DA. Born to be less equal: the predicament of the girl child in Tanzania. In: Gender, family and work in Tanzania. London: Routledge; 2018. p. 292–314.
Chapter Google Scholar
Opini B, Onditi H. Education for all and students with disabilities in Tanzanian primary schools: challenges and successes. Int J Educ Stud. 2016;3(2):65–76.
Pak K, Desimone LM, Parsons A. An integrative approach to professional development to support college-and career-readiness standards. Educ Policy Anal Arch. 2020;28(111): n111.
Palmary I. Back2School gender mainstreaming guidelines. 2024.
Prasetyo P, Azwardi A, Kistanti N. Gender equality and social inclusion (GESI) and institutions as key drivers of green entrepreneurship. Int J Data Netw Sci. 2023;7(1):391–8.
Shelley J. Identifying and overcoming barriers to gender equality in Tanzanian schools: educators’ reflections. Int J Pedagog Innov New Technol. 2019;6(1):9–27.
Siddaway AP, Wood AM, Hedges LV. How to do a systematic review: a best practice guide for conducting and reporting narrative reviews, meta-analyses, and meta-syntheses. Annu Rev Psychol. 2019;70(1):747–70.
Stoilova D, Cai R, Aguilar-Gomez S, Batzer NH, Nyanza EC, Benshaul-Tolonen A. Biological, material and socio-cultural constraints to effective menstrual hygiene management among secondary school students in Tanzania. PLOS Global Public Health. 2022;2(3): e0000110.
Thomas MA, Rugambwa A. Equity, power, and capabilities: constructions of gender in a Tanzanian secondary school. Fem Form. 2011;23(3):153–75.
Tieng’o EWB. Community perception on public primary schools: implications for sustainable fee free basic education in Rorya district, Tanzania. East Afr J Educ Soc Sci. 2019;1(1):32–47.
Wapling L. Inclusive education and children with disabilities: quality education for all in low and middle income countries. 2016. https://eajess.ac.tz/2020/05/26/community-perception-on-public-primary-schools-implications-for-sustainable-fee-free-basic-education-in-rorya-district-tanzania/ .
Zacharia L. Factors causing gender inequality in education in Tanzania: a case of Korogwe district secondary schools, PhD thesis. The Open University of Tanzania; 2014.
Download references
Author information
Authors and affiliations.
Tanzania Institute of Accountancy, Mwanza, Tanzania
Mbeya University of Science and Technology, Mbeya, Tanzania
Isack Kibona
You can also search for this author in PubMed Google Scholar
Contributions
H.E was collecting the literatures and read and write major parts I.K was good on drafting conclusion and analysis part. But we work hand on hand together.
Corresponding author
Correspondence to Henry Nkya .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/ .
Reprints and permissions
About this article
Nkya, H., Kibona, I. Systematic literature review of gender equity and social inclusion in primary education for teachers in Tanzania: assessing status and future directions. Discov Educ 3 , 122 (2024). https://doi.org/10.1007/s44217-024-00221-8
Download citation
Received : 26 March 2024
Accepted : 02 August 2024
Published : 13 August 2024
DOI : https://doi.org/10.1007/s44217-024-00221-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Equitable education
- Inclusive education
- Targeted intervention
- Find a journal
- Publish with us
- Track your research

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Clinicoecon Outcomes Res
- PMC11146608
Oral Prostacyclin Pathway Agents Used in PAH: A Targeted Literature Review
Charles d burger.
1 Division of Pulmonary Medicine, Mayo Clinic Florida, Jacksonville, FL, USA
2 Janssen Scientific Affiars, Titusville, NJ, USA
Marie Chivers
3 Avalere Health, Fleet, UK
Riya Vijay Vekaria
Gurinderpal doad.
4 Actelion Pharmaceuticals US, Inc, A Johnson and Johnson Co., Titusville, NJ, USA
Nikki Atkins
Sumeet panjabi.
Pulmonary arterial hypertension (PAH) is a rare and progressive pulmonary vascular disease that can result in right heart failure and death. Oral prostacyclins play an important role in the management of intermediate-low risk PAH. This targeted literature review (TLR) aimed to identify and compare evidence supporting use of oral prostacyclin pathway agents (PPAs: selexipag and oral treprostinil) in intermediate-low risk PAH.
A targeted literature review was conducted. Literature databases (MEDLINE, Embase, and Cochrane reviews) were searched for studies describing clinical practice and treatment outcomes for oral treprostinil and selexipag globally, published in English (2012 to 2022). Electronic searches were supplemented by manual-searches of targeted conferences (2020 to 2022), and reference lists of identified publications were reviewed. One reviewer assessed studies for eligibility.
In total, 95 publications met inclusion criteria: 47 full-text articles (selexipag n = 22; oral treprostinil n = 16; selexipag and oral treprostinil n = 9) and 48 conference materials. Selexipag and oral treprostinil target the prostacyclin pathway differently; their label-supporting trials had different primary endpoints (disease progression and hospitalization vs exercise capacity and disease progression), differing baseline therapy (0, 1 or 2 vs 0 or 1 baseline treatments), titration duration and dosing (personalized dose capped at 1600 ug twice daily (BID) vs increasing doses over time with no maximum dose), respectively. While both oral PPAs have demonstrated reduced risk of disease progression, only selexipag showed reduction in hospitalization rates. Oral PPAs have been shown to reduce healthcare costs in real-world clinical practice. This difference is reflected in labeled indications.
Given differences in trial- and real-world outcomes, number of prior therapies, and dosing, personalizing the choice of oral PPA is critical to maximizing the benefit for individual patients.
Plain Language Summary
PAH is a condition that causes heart failure. It is important to take medicines to slow down this process. For people with early disease, there are some medicines that can be taken as a tablet rather than as an injection to slow down disease progression. The differences between two of the tablet options – selexipag and oral treprostinil, are unclear. We reviewed publications describing how, when and why these medicines are used and how well they work, to improve our understanding of the value of these medicines to people with PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a rare disease resulting from progressive pulmonary vascular remodeling and luminal narrowing. 1 The typical symptoms of PAH (shortness of breath with exercise, fluid retention, lower extremity edema, and presyncope/syncope) are related to progressive decline in right heart function. 2 Changes in cardiopulmonary function often occur prior to symptom presentation in PAH. 3 The initial symptoms are often misdiagnosed as other cardiovascular and pulmonary conditions such as asthma or congestive heart failure, resulting in an average 2.8-year delay between symptom onset and diagnosis/treatment. 4 PAH is associated with high levels of morbidity and hospitalization burden. 5 Treatment is required to prevent right heart failure and death. 6 Treatment decisions are based on risk categorization, with initial double combination treatment targeting the endothelin and nitric oxide pathways recommended for low- or intermediate-risk patients; 6 however, only a small minority of patients maintain low-risk status following double combination treatment at 3-month follow-up. 7 The 2022 Guidelines of the European Society of Cardiology (ESC) and European Respiratory Society (ERS) describe the addition of a prostacyclin pathway agent, selexipag for patients on PDE5i and/or endothelin receptor antagonist (ERA) and oral treprostinil for patients on monotherapy (phosphodiesterase 5 inhibitor [PDE5i] or ERA) who are at risk of progression. 6
The prostacyclin metabolic pathway is dysregulated in patients with PAH, and PPAs play a key role in managing this condition; PPAs can be intravenous, subcutaneous, inhaled, or oral. 6 Treprostinil, a prostacyclin analog, and selexipag, a selective prostaglandin I2 receptor agonist, are the only two oral PPAs approved by the Food and Drug Administration (FDA) and represent a patient–friendly dosage alternative. 8 , 9 Oral treprostinil is not approved for use in PAH by the European Medicines Agency (EMA), whereas selexipag has been approved by the EMA. Earlier utilization of PPAs may improve long-term outcomes for PAH patients, delaying disease progression. Oral PPAs may improve uptake of PPAs by creating an easily accessible dosage option; however, information describing their use is diffuse. A review of the literature is needed to consolidate the evidence supporting use of the oral PPAs selexipag and treprostinil in clinical practice, to inform clinical and formulary decisions.
PAH is a rare disease, and many clinical studies in this indication are conducted in small patient populations. PAH describes a heterogeneous patient population comprising of patients at varying stages of progression and differing etiologies, and these differences are associated with impacts on clinical outcomes. Furthermore, there are differences in clinical trial endpoints between pivotal studies and differences in how morbidity endpoints are defined. There are challenges when performing meaningful indirect treatment comparisons between clinical trials in this indication for the reasons outlined. We performed a targeted literature review (TLR) to identify and compare all of the published data describing use of the oral PPAs treprostinil and selexipag (including the pivotal trials FREEDOM and GRIPHON, respectively) in PAH. The TLR identified clinical studies, prescribing practices, and clinical and economic outcomes for oral treprostinil and selexipag.
The TLR involved searching literature databases (MEDLINE, Embase, and Cochrane reviews) for articles describing clinical practice and treatment outcomes for oral treprostinil and selexipag in PAH. Aggregate search terms describing PAH, treprostinil, selexipag, reviews, real-world studies, and clinical trials were employed (see Table 1 for PICOS inclusion and exclusion criteria). The search was global and limited to publications in English from January 1, 2012, to June 23, 2022, including publications from targeted conferences. Targeted conferences (American College of Cardiology, American College of Chest Physicians, American College of Rheumatology, American Thoracic Society, British Society of Rheumatology, European Respiratory Society, European Society of Cardiology, International Society for Heart and Lung Transplantation, Pulmonary Hypertension Association, Pulmonary Vascular Research Institute) from 2020 to 2022 were reviewed. Article title and abstract (screen 1) and fulltexts (screen 2) were screened by a single reviewer, followed by a quality check conducted by a second independent reviewer. Studies in populations less than n = 20 were excluded from this analysis. The literature search was supplemented by a review of reference lists. A single reviewer screened article titles and abstracts (screen 1) and full texts (screen 2), followed by a quality check conducted by a second independent reviewer. Studies in populations of fewer than 20 people were excluded from this analysis.
PICOS Inclusion and Exclusion Criteria for the TLR
| Inclusion criteria | Exclusion criteria | |
|---|---|---|
| Population | ||
| Intervention | Oral treprostinil; Selexipag | |
| Comparator | Any/none | |
| Outcomes | Any clinical outcomes: specific outcomes of interest include mortality, PRO/quality of life, costs, treatment patterns, resource utilization, and prescriber’s behaviors, patient or carer experience | |
| Study design | ||
| Location | No country-specific restrictions | |
| Time | Limited to the last 10 years (2012–2022) | |
Notes : Conference search limited to: American College of Cardiology, American College of Chest Physicians, American College of Rheumatology, American Thoracic Society, British Society of Rheumatology, European Respiratory Society, European Society of Cardiology, International Society for Heart and Lung Transplantation, Pulmonary Hypertension Association, Pulmonary Vascular Research Institute.
Abbreviations : PAH, pulmonary arterial hypertension; PICOS, population, intervention, comparator, outcomes, study; PH, pulmonary hypertension; PRO, patient-reported outcome; RCT, randomized controlled trial; TLR, targeted literature review.
As illustrated in the PRISMA diagram illustrated in Figure 1 , 992 articles were identified through MEDLINE, Embase, and Cochrane reviews; 7 additional articles were identified through review of conference materials and a further 12 articles were identified through a review of reference lists. One hundred and ninety-three articles progressed to full content screening with 95 publications meeting the inclusion criteria for extraction: 48 conference materials, 47 full-text articles (selexipag n = 22; oral treprostinil n = 16; selexipag and oral treprostinil n = 9). Most studies identified were observational and clinical studies describing dose titration and safety. No studies were identified that described unmet need, treatment patterns, patient or physician preferences, or patient and caregiver experiences.

PRISMA diagram.
The findings from this TLR confirmed the diffuse nature of the literature describing outcomes for these oral PPA treatments. Half of the literature identified had been published within conference materials. Many of the studies identified were in small patient numbers and reflected the heterogeneous nature of these patient populations (both stage of disease progression and etiology).
Randomized Controlled Trials (RCTs) and FDA Indications for Selexipag and Oral Treprostinil
Primary endpoints and outcomes of the RCTs of selexipag and oral treprostinil are summarized in Table 2 . Several studies have suggested that PAH-related morbidity events are prognostic for mortality. 10 , 11 Clinical trials of selexipag (GRIPHON) and oral treprostinil (FREEDOM-EV) have used composite endpoints to measure disease progression as a primary endpoint. 11–14
Registrational Clinical Trials of Selexipag and Oral Treprostinil
| GRIPHON (Sitbon et al, 2015) | FREEDOM-EV (White et al, 2020) | FREEDOM-M (Jing et al, 2013) | FREEDOM-C (Tapson et al, 2012) | FREEDOM-C2 (Tapson et al, 2013) | |
|---|---|---|---|---|---|
| Selexipag | Oral treprostinil | Oral treprostinil | Oral treprostinil | Oral treprostinil | |
| 5 years | 6 years | 12 weeks | 16 weeks | 16 weeks | |
| Selex = 574; Plb = 582 | Trep = 346; Plb = 344 | Trep = 233; Plb = 116 | Trep = 174; Plb = 176 | Trep = 157; Plb = 153 | |
| 46% FC II; 53% FC III | 63% FC II; 34% FC III | 36% FC II; 61% FC III | 21% FC II; 76% FC III | 26% FC II; 73% FC III | |
| IPAH 56.1% CTD 28.9% CHD 9.5% | NR | IPAH 73% | IPAH 66% CTD 28% | IPAH 66% CTD 31% CHD 2% | |
| 80% | 100% | 0 | 100% | 100% | |
| 20% | 0 | 0 | 0 | ||
| 47% | 100% | 55% | 60% | ||
| 33% | 0 | 45% | 40% | ||
| Time to first morbidity/mortality event (composite) | Clinical worsening event (composite) | Change from baseline in 6MWD at week 12 | Change from baseline in 6MWD at week 16 | Change from baseline in 6MWD at week 16 | |
| 40% risk reduction) | 25% risk reduction | 23.0 m vs Plb | 11 m vs Plb (P = 0.07) | 10.0 m vs Plb (P = 0.089) | |
| 200–1600 µg BID | 0.125–12 mg TID 3.56 mg TID at week 24 (median) | 0.25–12 mg BID 3.6mg BID at week 12 (mean) | 0.25–16 mg BID 3.0mg BID at week 16 (median) | 3.1 mg BID (mean) | |
| 14.3% | 18.8% | 9.9% | 14.4% | 11.5% | |
| H (65% vs 33%), D (42% vs 19%), N (34% vs 19%), jaw pain (26% vs 6%) | H (70%), D (66%), flushing (44%), N (37%) | H (69%), N (39%), D (37%), pain in jaw (25%) | H (86%), N (64%), D (61%), flushing (49%) | H (71%), D (55%), N (46%), flushing (35%) |
Notes : This table represents the pivotal/registrational trials for these oral PPA agents but does not include all studies identified in the literature review.
Abbreviations : AE, adverse event; BID, twice a day; CHD, congenital heart disease; CTD, connective tissue disease; D, diarrhea; DC, discontinuation; FC, functional class; H, headache; IPAH, idiopathic pulmonary arterial hypertension; N, nausea; NR, not reported; PAH, pulmonary arterial hypertension; Plb, placebo; Selex, selexipag; TID, three times a day; Trep, treprostinil; 6MWD, 6-minute walking distance.
The FDA-approved indication for selexipag is for treatment of PAH (World Health Organization [WHO] Group 1) to delay disease progression and reduce the risk of hospitalization. 18 This reflects findings from GRIPHON, as selexipag significantly reduced the risk of morbidity and mortality events versus placebo by 40%, 13 and resulted in a 30% risk reduction for the secondary endpoint, risk of death or hospitalization due to PAH worsening, compared with placebo (hazard ratio [HR]: 0.70; 95% confidence interval [CI]: 0.54, 0.91); 87.4% of the events for this endpoint were PAH-related hospitalizations. 13 In GRIPHON, time from diagnosis was ≤6 months in 34.9% of patients and >6 months in 65.1% of patients. The value of earlier initiation of selexipag after diagnosis (≤6 months versus >6 months) has been established in a post hoc analysis of the GRIPHON trial; patients who initiated selexipag earlier experienced a more pronounced effect on the time to first disease progression event than those who initiated later (HR: 0.45; 95% CI: 0.33, 0.63, and HR: 0.74; 95% CI: 0.57, 0.96, respectively; P = 0.0219 for interaction). 19
Oral treprostinil is indicated by the FDA for the treatment of PAH (WHO Group 1) to delay disease progression and improve exercise capacity. 20 Oral treprostinil was studied in a series of randomized, prospective, placebo-controlled clinical trials (the “FREEDOM” trials). Six-minute walk distance (6MWD) at week 12 was the primary endpoint in the FREEDOM-M trial. It showed a significant benefit compared with baseline values (median Hodges-Lehmann treatment effect of 23.0m [95% CI, 4–41 m; P = 0.0125]), leading to FDA approval of oral treprostinil for use as a monotherapy to improve exercise capacity in PAH. 15 Trials of oral treprostinil added to background therapy (FREEDOM-C and FREEDOM-C2) failed to demonstrate a significant improvement in the primary endpoint, 6MWD. 16 , 17 The primary endpoint of FREEDOM-EV was time to first adjudicated clinical worsening event. In FREEDOM-EV, 90 (26%) participants in the oral treprostinil group experienced a clinical worsening event compared with 124 (36%) of placebo participants (HR: 0.74; 95% CI: 0.56, 0.97; P = 0.028). The treatment-attributable difference in clinical worsening was driven by reduced incidence of disease progression in the oral treprostinil group (HR 0.39; 95% CI 0.23, 0.66; P < 0.001). 14 Findings from FREEDOM-EV expanded the oral treprostinil FDA label to include delay in disease progression. 20
Survival Data
GRIPHON was not powered to determine survival outcomes, and no statistical difference in the number of deaths among patients receiving oral selexipag (17.4%) was observed versus placebo (18.0%). 13 GRIPHON mortality data are difficult to interpret because of treatment switching following a non-fatal primary endpoint event, with many patients switching from placebo to oral selexipag. 13 Post hoc analysis of the FREEDOM-EV study found that mortality was lower at study closure in patients receiving oral treprostinil versus placebo (11% vs 17.4%, respectively; P = 0.0324). 21 However, this finding is difficult to interpret because mortality was similar between oral treprostinil and placebo at the end of randomized treatment (4.9% vs 5.2%, respectively, P = 0.9781) or open-label extension study (8.7% vs 12.2%, respectively, P = 0.43), and death status was unknown for 11% of the original FREEDOM-EV population. 21
Survival outcomes in longer term open-label studies for progressive conditions such as PAH can be impacted by variation in subsequent treatments. Long-term follow-up of patients who received selexipag in the placebo-controlled GRIPHON study and the open-label extension study showed that Kaplan–Meier estimates of overall survival at 1, 2, 5, and 7 years were 92%, 85%, 71%, and 63%, respectively. 22 Long-term follow-up of patients who received oral treprostinil in the placebo-controlled FREEDOM-EV and open-label extension study showed Kaplan–Meier estimates of overall survival at 1, 3, and 5 years were 96%, 88%, and 79% versus 95%, 80%, and 70% for placebo, respectively. 23 It is not possible to directly compare survival data between GRIPHON and FREEDOM-EV due to differences in background therapy (ie, 32.5% of patients in GRIPHON received dual-combination PAH treatment, while all patients in FREEDOM-EV received PAH monotherapy); 19 , 21 and function class with a greater proportion of FC III patients in GRIPHON compared with FREEDOM-EV (52.5% vs 33.9% respectively).
Mortality outcomes are difficult to determine for oral PPAs due to the absence of comparative data, 22–24 and the short duration of clinical trials. 13–15
Place in Therapy
Based on 2022 ESC/ERS guidelines, initial triple oral combination therapy with oral PPAs is not recommended but plays a role for patients who present at intermediate-low risk of death while receiving ERA or PDE5i therapy. 6 Sequential triple therapy with selexipag is supported by post hoc analysis of patients on dual combination background therapy in GRIPHON. 24 TRITON assessed initial triple combination with selexipag versus initial dual combination with selexipag over 26 weeks; although no significant difference in pulmonary vascular resistance was observed, exploratory analyses suggested a possible signal for improved long-term outcomes. 25 Data supporting the use of oral treprostinil in triple combination therapy is lacking; additional studies will be required to evaluate the efficacy of adding oral treprostinil to dual combination therapy (see Table 2 ). 16 , 17
As illustrated in Figure 2a , real-world evidence indicates that, overall, oral selexipag is predominantly used within a triple combination regimen (31.0% to 88.0% of patients receiving selexipag); 26 , 27 newly diagnosed patients more often initiate selexipag treatment with a dual combination regimen with an ERA or a PDE5i. 26 As illustrated in Figure 2b , oral treprostinil is equally used as a monotherapy or within a dual/triple-combination regimen with an ERA and/or PDE5i. Studies from the ADAPT registry describe oral treprostinil use within triple combination (33.3% to 45.7% of patients receiving oral treprostinil). 28 , 29 However, neither of the early oral treprostinil trials, FREEDOM-C and FREEDOM-C2, demonstrated an improvement in 6MWD with the addition of oral treprostinil to double oral combination therapy (see Table 2 ). 16 , 17

Real-world evidence studies describing proportions of patient using selexipag and oral treprostinil within a combination regimen. ( a ) illustrates the proportions of patients taking selexipag as a monotherapy or within dual- or triple-combination regimens in reported studies; ( b ) illustrates the proportions of patients taking oral treprostinil as a monotherapy or within dual- or triple-combination regimens in reported studies. a This value has been calculated based on patient baseline characteristics, assuming the background therapy has been supplemented with one additional therapy rather than replaced.
Dosing and Titration of Oral PPAs
As summarized in Table 3 , both oral PPAs require initial titration. Individualized dose titration is required for oral PPAs to identify an upper maintenance dose that avoids unmanageable side effects. 31
Selexipag and Oral Treprostinil Dosing in RCTs and Clinical Practice
| Selexipag | Oral Treprostinil | ||||
|---|---|---|---|---|---|
| GRIPHON a | Up-titration by 200 µg BID per week until maintenance dose reached; if AEs occur, decrease by 200 µg in both daily doses Week 12: 23% of patients at 200–400µg BID; 31% of patients at 600–1000µg BID; 43% of patients at 1200–1600µg BID | Freedom-M c | Initiated 1.0 mg BID; dose escalation 0.25 to 0.5 mg BID every 3 days to maximum of 12 mg BID Week 12: 3.6 mg BID = 7.2 mg TDD | ||
| Freedom-C b | Initiate 0.5 mg BID; changed to 0.25 mg BID Week 16: 3.0 mg BID = 6.0 mg TDD | ||||
| Freedom-C2 c | Patients on background ERA and/or PDE5i initiated 0.25 mg BID; dose escalation 0.25 mg BID for 3 days; after 4 weeks dose escalations of 0.25 or 0.5 mg BID every 3 days Week 16: 3.1 mg BID = 6.2 mg TDD | ||||
| Freedom-Ext b | Year 1: 7 mg TDD Year 2: 8 mg TDD Year 3: 8.25 mg TDD | ||||
| Freedom-EV b | Daily up-titration in 0.125 mg increments for first 4 weeks then 0.25 mg daily to maximum of 12 mg (TID dosing with food) Week 24: Median 3.56 mg TID = 10.68 mg TDD | ||||
| SPHERE Registry b (n = 500) | 1200 µg BID Median time to maintenance dose: 8.1 weeks | El-Kersh et al, 2020 b (n = 2255) | TID dosing (85% patients, n = 1917) Month 3: 5.1 mg TDD Month 6: 8.4 mg TDD Month 12: 10.8 mg TDD Month 18: 12.0 mg TDD Month 24: 12.2 mg TDD Month 36: 12.9 mg TDD | BID dosing (15% patients, n = 338) Month 3: 2.3 mg TDD Month 6: 4.3 mg TDD Month 12: 5.4 mg TDD Month 18: 5.4 mg TDD Month 24: 5.4 mg TDD Month 36: 6.4 mg TDD | |
| Kung et al, 2012 c (n = 2490) | Day 83 (~12 weeks): 15.6% patients at 200–400 µg BID; 33.9% patients at 600–1000 µg BID; 50.5% patients at 1200–1600 µg BID | ||||
| N/A | Rahaghi et al, 2017 b | Slow titration in 0.125 mg dose increments with 6–8 hours between doses in “stair-step” titration Month 3: Target dose: 4 mg TID = 12 mg TDD Month 6: Target dose:6 mg TID = 18 mg TDD Month 12: Target dose: 8 mg TID = 24 mg TDD | |||
Notes : a Percentage of patients at each dose stratification level; b median dose; c mean dose; d median time to individualized maintenance dose was measured in the first 500 patients; e TDD was calculated by multiplying individual dose by dosing frequency.
Abbreviations : AE, adverse event; BID, twice a day; ERA, endothelin receptor antagonist; N/A, not applicable; PDE5i, phosphodiesterase 5 inhibitor; RCT, randomized controlled trial; TDD, total daily dose; TID; three times a day.
Selexipag Titration and Dosing
GRIPHON describes the titration regimen for selexipag of 200 µg BID increased in increments of 200 µg BID weekly until side-effects cannot be managed by adequate treatment or to the maximal dose of 1600 µg BID. Across all doses, selexipag demonstrated a reduction in the risk of morbidity and mortality, supporting individual-dose titration. For patients experiencing a prostacyclin-associated side effect that is unmanageable, the dose is reduced by 200 µg. 13
Real-world evidence (SPHERE registry) suggests that most patients (87.8%) receiving selexipag titrate more slowly than the label recommended 200 µg BID weekly, with over one-fifth of patients (21.6%) titrating at 200 µg BID every two weeks. 32 The majority of studies that reported selexipag titration described completing titration over 7–9 weeks; 33 , 39 , 40 SPHERE registry data reported a median time from selexipag initiation to an individualized maintenance dose of 8.1 weeks, 32 , 40–42 and a median maintenance dose of 1200 µg BID (n = 496). 40 SPHERE registry data indicate that the average maintenance dose continued to increase and began to stabilize at 6 months. At 18 months, 77.6% of patients enrolled in SPHERE completed the study; of the 22.4% who discontinued early, 11.4% discontinued due to adverse events (AEs) related to PAH progression and 11.2% discontinued due to AEs not related to progression (7.2% attributable to selexipag). 32
Oral Treprostinil Titration and Dosing
Across the FREEDOM trials, oral treprostinil titration and dosing evolved with variation in target doses ( Table 3 ). These variations arose from changes in the available tablet strength, adoption of three times daily (TID) versus BID dosing, and expert panel recommendations. 15–17 , 20 The FREEDOM-EV trial describes a higher dose than earlier trials, with daily up-titration in 0.125 mg increments for the first 4 weeks, then 0.25 mg increments daily to a maximum of 12 mg (TID dosing with food), achieving an upper median dose of 3.56 mg TID by week 24. 23
The minimum effective dose for oral treprostinil is not clear from the literature; however, the literature indicates a dose-dependent treatment effect.
- An open-label study of patients participating in FREEDOM trials followed 37 patients and reported mean oral treprostinil doses of 4.3 ± 2.3 to 11.7 ± 5.8 mg/24 hours between 3 and 24 months. Oral treprostinil dose was inversely associated with a change in pulmonary vascular resistance (r = −0.42, P < 0.05), with a greater change among patients in the highest dosing quartile. 43
- Analysis of oral treprostinil dose and response in 1619 patients with PAH indicated that higher doses of oral treprostinil are required to achieve significantly longer times for first PAH-related and all-cause hospitalization; a trend towards improvements in 6MWD was observed with higher doses. 44
The target dose across FREEDOM studies varied, as reflected in real-world studies ( Table 3 ). A study using specialty pharmacy service shipment records indicated that in the overall TID dosing group (n = 1200), the median total daily dose (TDD) varied from 5.3 mg to 10.8 mg between 3 and 18 months. In the BID dosing group (n = 400), median TDD increased from 2.5 mg to 5.5 mg between 3 and 18 months. 45 More prevalent use of TID has improved tolerability, leading to higher TDDs.
Twenty-two studies were identified reporting safety findings for selexipag and 8 studies for oral treprostinil.
The most commonly reported AEs for oral selexipag in GRIPHON were headache (65%), diarrhea (42%), nausea (34%), pain in jaw (26%), and vomiting (18%) (Table 2). 13 , 15 In FREEDOM-M, FREEDOM-C, and FREEDOM-C2 OLE, the most frequently reported AEs were headache (71%), diarrhea (55%), nausea (46%), flushing (35%), vomiting (21%), and pain in jaw (25%). 17 Findings in FREEDOM-EV were similar. 21
EXPOSURE (observational study) found that selexipag maintenance treatment at the individualized dose was well tolerated in clinical practice. 46 A registry study of oral treprostinil found that the rate of AEs decreased over time, with a large reduction in reported rates of prostacyclin-related AEs by month 6. 30
Effects of Oral PPAs on Healthcare Utilization and Costs (Oral Selexipag versus Oral Treprostinil)
PAH is characterized by frequent hospitalizations and high medical costs. 5 Eight studies were identified that describe economic outcomes for selexipag, including two comparing outcomes with oral treprostinil. No head-to-head clinical trials have compared the impact of these two oral PPAs on hospitalization.
- A retrospective claims analysis of 222 people in the US compared outcomes in patients receiving oral PPAs; compared with oral treprostinil (n = 99), selexipag (n = 123) was associated with a 46% lower risk of all-cause hospitalization (P = 0.02) and a 47% lower risk of pulmonary hypertension (PH)-related hospitalization (P = 0.03). 47
- In a retrospective cohort study using the IBM MarketScan database, Dean et al studied patients aged ≥18 years receiving selexipag (n = 126) or oral treprostinil (n = 130). At month 6, the mean number of inpatient visits was similar between treatment groups (adjusted all-cause inpatient visits: 1.3 ± 0.6 [oral treprostinil] vs 1.9 ± 0.8 [selexipag] (P = 0.2); 48 and adjusted PAH-related inpatient visits: 1.2 ± 0.6 vs 1.2 ± 0.6, respectively; P = 1.0). After adjusting for covariates, treatment with selexipag was associated with 51.4% higher total all-cause healthcare costs versus oral treprostinil, predominantly attributable to all-cause pharmacy costs (68.2% higher in patients receiving selexipag compared with those receiving oral treprostinil). 48 , 49
- A retrospective cohort study of 1,310 patients with PAH (using the Optum database) reported all-cause hospitalization costs of US$39,983 for oral treprostinil versus US$20,635 for selexipag. Total PAH-related medical costs were 40% lower (US$4,03,987) for patients receiving selexipag versus oral treprostinil (P = 0.006). 50
- A retrospective claims analysis of 583 patients in the US reported that after adjustment for baseline characteristics, selexipag had the lower risk for hospitalization when compared to inhaled iloprost and parenteral treprostinil (relative rate ratio [95% CI], 0.40 [0.22, 0.75], and 0.26 [0.17, 0.39]) and outpatient visits (0.66 [0.56, 0.78] and 0.76 [0.66, 0.88], respectively). 34
Although oral treprostinil and selexipag both target the prostacyclin pathway, these medications are not clinically equivalent and have different treatment effects on specific health outcomes (eg, hospitalization) and further differences in safety profiles, titration, and maintenance management. Efficacy is also variable depending on the number of concomitant PAH therapies (one vs two) and dose level (individualized dose vs need to maximize dose). Population health decision-makers should strive to ensure that all PAH therapies are made available for patients given the risk of severe negative outcomes, including hospitalization and death in patients with poorly controlled disease, enabling expert clinicians to individualize therapy, including the choice of PPAs based on their experience, available evidence, and guidelines. To optimize outcomes with oral PPAs, careful management of initial side-effects is essential to complete titration and initiate maintenance treatment successfully. Patient tolerability is likely to improve, as side-effects are reported to decrease over time. 44
The cost for selexipag is fixed and predictable as it does not vary by dose. Oral treprostinil cost is subject to variation by dose, with increasing costs associated with higher maintenance doses, and so it is difficult to predict and model annual costs incurred with this treatment. Treatment costs should not be viewed in isolation for oral PPAs, as this is a rare fatal disease associated with significant burden on patients and the healthcare system. Oral PPAs deliver many economic benefits, including reduced healthcare costs, reduced hospitalization rates (selexipag), and delayed disease progression, offsetting costs associated with use of these medications.
Prior authorizations, step-edits, and quantity limits are common for oral PPAs due to the high treatment costs. Utilization management strategies that are too restrictive may delay initiation of therapy, resulting in poorer outcomes. Indeed, prior authorizations for life-saving medications may be a misplaced strategy when the delay can lead to worsening outcomes or incremental costs, or both. Prompt PPA therapy initiation has been associated with improved or stabilized clinical status for this rapidly progressing disease with a high mortality rate. 19
ESC/ERS guidelines recommend risk assessment every 3 to 6 months, which serves as an early signal for treatment escalation with an oral PPA. Population health decision-makers may implement risk assessments to detect patients at risk of worsening and treatment escalation.
Limitations
Studies conducted on fewer than 20 patients were not included in the evidence synthesis. The heterogeneity of clinical trial design (endpoint definition, patient population – etiology, stage of disease progression) are limitations when comparing outcomes across the clinical studies describing in this literature review. The findings of the literature review reflect publications up to June 2022 and do not include subsequent publications.
Conclusions
The 2022 ESC/ERS guidelines describe the addition of selexipag for patients receiving PDE5i and/or ERA and oral treprostinil for patients receiving monotherapy (PDE5i or ERA) who are at risk of progression. This TLR provides population health decision-makers with important insights to evaluate the distinct profiles of oral PPA treatment options and to inform formulary and coverage decisions for the treatment of patients, with PAH ensuring access to critical PAH therapies.
Funding Statement
This study was funded by Janssen USA and conducted in partnership with Avalere Health, UK. Janssen provided input into the initial study concept and design and Avalere Health led the study execution with input and review from the authoring team.
Dr Charles Burger reports personal fees from Janssen, personal fees from INSMED, personal fees from Merck, and non-financial support from United Therapeutics, during the conduct of the study. Yuen Tsang is a former employee and owns stock in Johnson & Johnson. Dr Marie Chivers, Nikki Atkins and Ms Riya Vekaria report employment with Avalere Health which was in receipt of payment from Janssen for this element of the study. Dr Gurinderpal Doad and Dr Sumeet Panjabi are employees and stockholders of Johnson & Johnson, the company that markets oral selexipag. The authors report no other conflicts of interest in this work.
- Open access
- Published: 12 August 2024
Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search
- Dongye He ORCID: orcid.org/0000-0001-6704-7354 1 , 2 ,
- Mei Zhang 1 , 3 ,
- Yanying Li 1 , 3 ,
- Fupeng Liu 1 , 2 &
- Bo Ban ORCID: orcid.org/0000-0002-3950-1422 1 , 2 , 3
Orphanet Journal of Rare Diseases volume 19 , Article number: 292 ( 2024 ) Cite this article
27 Accesses
Metrics details
Ankyrin repeat domain containing-protein 11 (ANKRD11), a transcriptional factor predominantly localized in the cell nucleus, plays a crucial role in the expression regulation of key genes by recruiting chromatin remodelers and interacting with specific transcriptional repressors or activators during numerous biological processes. Its pathogenic variants are strongly linked to the pathogenesis and progression of multisystem disorder known as KBG syndrome. With the widespread application of high-throughput DNA sequencing technologies in clinical medicine, numerous pathogenic variants in the ANKRD11 gene have been reported. Patients with KBG syndrome usually exhibit a broad phenotypic spectrum with a variable degree of severity, even if having identical variants. In addition to distinctive dental, craniofacial and neurodevelopmental abnormalities, patients often present with skeletal anomalies, particularly postnatal short stature. The relationship between ANKRD11 variants and short stature is not well-understood, with limited knowledge regarding its occurrence rate or underlying biological mechanism involved. This review aims to provide an updated analysis of the molecular spectrum associated with ANKRD11 variants, investigate the prevalence of the short stature among patients harboring these variants, evaluate the efficacy of recombinant human growth hormone in treating children with short stature and ANKRD11 variants, and explore the biological mechanisms underlying short stature from both scientific and clinical perspectives. Our investigation indicated that frameshift and nonsense were the most frequent types in 583 pathogenic or likely pathogenic variants identified in the ANKRD11 gene. Among the 245 KBGS patients with height data, approximately 50% displayed short stature. Most patients showed a positive response to rhGH therapy, although the number of patients receiving treatment was limited. ANKRD11 deficiency potentially disrupts longitudinal bone growth by affecting the orderly differentiation of growth plate chondrocytes. Our review offers crucial insights into the association between ANKRD11 variants and short stature and provides valuable guidance for precise clinical diagnosis and treatment of patients with KBG syndrome.
The ANKRD11 gene (OMIM#611192) is mapped to human chromosome 16q24.3 and encodes an ankyrin repeat domain-containing protein 11 that belongs to a member of the ankyrin repeats-containing cofactor family (ANCO). It is relatively conserved across species and ubiquitously expressed in multiple organs and tissues, particularly in the brain and ovary [ 1 , 2 ]. The ANKRD11 protein, consisting of 2,663 amino acid residues, structurally includes the ankyrin domain (ANK), transcriptional activation domain (AD), transcriptional repression domains (RD1 and RD2), and multiple putative nuclear localization signals (NLSs) [ 3 ]. The N-terminal ANK domain follows the canonical helix-loop-helix-β-hairpin/loop configuration and is comprised of five consecutive ankyrin repeat motifs. Each motif contains a 33-residue sequence and facilitates protein-protein interaction to coordinate subsequent transcriptional regulatory processes [ 4 , 5 , 6 ]. The ANKRD11 protein binds to the conserved N-terminal Per-Arnt-Sim (PAS) region of p160 coactivator via its ANK domain, concurrently, recruits histone deacetylases (HDACs) through its RD1 or RD2 domain. When p160 coactivator binds to the hydrophobic cleft within the C-terminal ligand-binding domain (LBD) of nuclear receptors (NRs) through its LXXLL motifs, the assembly of p160/ANKRD11/HDACs complex suppresses NRs-mediated ligand-dependent transactivation [ 7 ]. The ANKRD11 protein also interacts with the N-terminal 84 amino acids of ADA3 (alteration/deficiency in activation 3), which is an essential part of the p300/CBP [cAMP-response-element binding protein-binding protein]-associated factor (P/CAF) complex. This complex connects coactivators to histone acetylation and basal transcription machinery, resulting in the recruitment of the P/CAF complex and the specific regulation of ADA3 coactivator in a transcription factor-dependent manner [ 8 ]. Moreover, the ANKRD11 protein is capable of amplifying p53 activity through the enhancement of P/CAF-mediated acetylation [ 6 ]. Overall, the ANKRD11 protein, through its various functional domains, collectively facilitates the formation of a molecular bridge between coactivators or corepressors and histone deacetylases (HDACs) or histone acetyltransferases (HATs), thereby precisely regulating the transcription of target genes.
Initially, ANKRD11 has been recognized as a tumor suppressor gene in breast cancer due to its location within the chromosomal region 16q24.3, which is widely acknowledged for its frequent loss of heterozygosity (LOH) among patients suffering from breast cancer [ 9 , 10 ]. Under normal physiological conditions, the estrogen receptor (ER)/amplified in breast cancer 1 (AIB1)/ANKRD11/HDACs or transcriptional enhanced associate domain (TEAD)/yes-associated protein (YAP)/AIB1/ANKRD11 complex functions to suppress the transcriptional activation of oncogenes in breast cancer [ 11 , 12 ]. However, aberrant DNA methylation of three CpGs within a 19-base pair region of the ANKRD11 promoter leads to its down-regulation, thereby disrupting the assembly of the complex and consequently promoting breast tumorigenesis [ 13 ]. ANKRD11 haploinsufficiency was later identified in KBG syndrome (KBGS) patient-focused clinical and molecular studies, confirming the dominant pathogenic mechanism responsible for this condition (OMIM#148050). KBGS was initially reported by Herrmann and colleagues in 1975 and characterized by macrodontia of the upper central incisors, distinctive craniofacial findings, postnatal short stature, skeletal anomalies and, neurodevelopmental disorders, sometimes with seizures and electroencephalogram (EEG) abnormalities [ 14 , 15 , 16 ]. Patients harboring ANKRD11 pathogenic variants exhibit overlapping features between KBGS and Cornelia de Lange syndrome or Coffin-Siris-like syndrome, particularly neurological and skeletal anomalies [ 17 , 18 ]. KBGS typically presents with a wide range of phenotypic manifestations, each varying in severity [ 19 ]. The biological function and cellular mechanism of ANKRD11 variants associated with the KBGS features have garnered significant interest and attention within the academic community. Previous study has established the pivotal role of the ANKRD11 gene in proliferation, neurogenesis and neuronal localization of cortical neural precursor cells by utilizing a Yoda mice model harboring a point mutation within the ANKRD11-HDAC interaction region, and the underlying mechanism was linked to alterations in the acetylation patterns of specific lysine residues (H3K9, H4K5, H4K8, H4K16) on the target genes regulated by ANKRD11 [ 20 ]. Further investigation has revealed that ANKRD11 regulates pyramidal neuron migration and dendritic differentiation of mouse cerebral cortex through the coordination of P/CAF to facilitate the acetylation of both p53 and Histone H3, which subsequently leads to the activation of brain-derived neurotrophic factor (BDNF)/tyrosine receptor kinase B (TrkB) signaling pathway [ 21 ]. Moreover, Roth and their colleagues developed a heterozygous neural crest-specific ANKRD11-mutant mice model, and revealed that multiple ossification centers in the middle facial bone of mice failed to expand or fuse properly, leading to a significant delay in bone maturation and a severe restriction in bone remodeling [ 22 ]. Recent research has uncovered that conditional knockout of the ANKRD11 gene within murine embryonic neural crest leads to severe congenital cardiac malformations and the underlying mechanism was linked to a reduction in Sema3C expression levels, coupled with diminished mTOR and BMP signaling within the cardiac neural crest cells of the outflow tract [ 23 ]. Based on the accumulating evidence from ongoing research into gene functions, the relationship between ANKRD11 pathogenic variants and the clinical features of KBGS is better understood than ever before. However, the role of ANKRD11 variants in inducing short stature has not received sufficient attention, particularly regarding its frequency of occurrence and the underlying biological mechanisms of action.
Materials and methods
We investigated publicly available online resources including published literature in Web of Science, PubMed, Google Scholar, and Wanfang database by searching keywords “KBGS”, “ANKRD11”, “Short stature” and “Intellectual disability” as well as genetic testing records in ClinVar database between July 2011 and March 2024. In this review, we included a total of 78 published papers that encompassed cohort studies, case series or single-case reports, and gathered 583 ANKRD11 variants, which were classified as pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG)-Association for Molecular Pathology (AMP) guideline (Supplemental material 1 ). Among these variants, 202 were reported in published papers and 381 were described in the ClinVar database. Certain large deletions or duplications of the ANKRD11 gene were not considered in this analysis, as the complexity of their impact on the amino acid sequence of the encoded protein posed challenges for interpretation. We have also excluded patients with 16q24.3 microdeletions, 16q24.3 microduplications and dual molecular diagnosis involving ANKRD11 and/or flanking genes, as the role of other genes in contributing to the height phenotype remains uncertain. Furthermore, hotspot variants within ANKRD11 were analyzed in 838 patients, comprising 457 derived from the literature and 381 derived from the ClinVar database (Supplemental material 2 ). ANKRD11 allele frequency below 1% in the general poulation was obtained from gnomAD ( http://gnomad-sg.org/ ). 245 patients were reported to have height data, of which 112 had a height SDS. The differences in height SDS among patients with short stature carrying various ANKRD11 variants were further analyzed (Supplemental material 3 ). Data was described as mean ± SDS, and analyzed with one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. A significant difference was considered when the p -value was less than 0.05.
Molecular spectrum of ANKRD11 variants
Since ANKRD11 was identified as the causal gene for KBGS in 2011, more than 340 KBGS patients have been reported worldwide [ 24 ]. Considering the variant data documented in the ClinVar database, it is projected that the number of patients with ANKRD11 variants exceeds 800. Despite the global prevalence of KBGS worldwide remaining unknown, its prevalence is underestimated due to a limited understanding of the disease phenotype and molecular underpinning. Consequently, establishing the spectrum of genetic variation in the ANKRD11 gene holds the promise of not only enhancing our understanding of disease’s pathogenesis but also enabling clinicians to render a precise molecular diagnosis for KBGS. A total of 583 ANKRD11 variants encompassed nearly the entire sequence of amino acids [ 1 , 2 , 15 , 17 , 18 , 19 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 ] (Fig. 1 ). All identified ANKRD11 variants were present in a heterozygous state, aligning with early embryonic lethality of Yoda mice observed in homozygotes, as demonstrated by Barbaric et al. [ 3 ]. This review encapsulates the up-to-date molecular landscape of ANKRD11 variants, nevertheless, in light of the continual discovery of patients with newly identified ANKRD11 variants, it needs to be supplemented and updated in time.

Molecular spectrum of ANKRD11 variants. A total of 583 ANRKD11 (likely) pathogenic variants were collected through literature review and ClinVar database. ANKRD11 variants were shown by frameshift, nonsense, missense, splice and inframe deletion, respectively. ANK: ankyrin repeat domain, RD1: repression domain 1, AD: activation domain, RD2: repression domain 2
All ANKRD11 variants in the map were classified into five types: frameshift variants (340/583, 58.32%), nonsense variants (163/583, 27.96%), missense variants (54/583, 9.26%), splicing variants (21/583, 3.60%) and inframe deletion variants (5/583, 0.86%) (Fig. 2 ). Variants occurring in ANK, RD1, AD, RD2 and non-domain region accounted for 3.60%, 10.12%, 9.09%, 15.10% and 62.09% of the total variant pool, respectively (Fig. 2 ). Multiple putative NLSs within the interval between the RD1 and AD regions were categorized as part of the non-domain segment, primarily due to the absence of definitive and evidence-based localization data [ 3 , 5 , 15 , 48 ]. Specific variants occurring within these NLSs may impair the nuclear targeting of the ANKRD11 protein. Notably, the most common variants were frameshift and nonsense variants, which give rise to prematurely truncated forms of the ANKRD11 protein. 62.96% (34/54) of ANKRD11 missense variants were found to cluster within C-terminal RD2 region. The majority of these missense variants, particularly those impacting arginine residues, were reported to impair protein stability or transcriptional activity, however, they did not produce an obvious impact on the protein’s subcellular localization [ 61 , 66 ]. Additionally, alternative splicing events predominantly affected the C-terminal RD2 (13/21) and N-terminal region (8/21). It is not surprising that those affecting 5’ and 3’ splice sites are commonly implicated as the underlying cause of hereditary disorders [ 97 ]. Nonetheless, how these hypothesized splicing variants impact the encoded protein requires an in-depth examination of splicing patterns by cDNA analysis, and frequently involves a Mini-gene assay. Other types of ANKRD11 variants were relatively uncommon including p.Lys1347del, p.Thr2471_Gly2474del, p.Glu2524_Lys2525del, p.Q2350del, and p.R595_A2663delinsS. Interestingly, p.Lys1347del has been demonstrated to significantly disrupt the transcriptional activation of downstream p21 gene but did not influence the levels of ANKRD11 mRNA or protein [ 2 , 15 , 19 , 61 ]. Theoretically, protein-truncating variants (PTVs) cause a more detrimental effect on protein function compared to the consequences of amino acid deletions (≥ 1) and single amino acid substitution [ 98 , 99 ]. The impact of various types of genetic variants on the ANKRD11 protein function requires further investigation by a range of functional analyses.

The percentage of different types of ANKRD11 variants located in different functional domains. The pie chart indicates the percentage of variants within different domains. 10 X 10 dot plot represents the percentage of different variant types. The column shows the the proportion of five mutation types within different domains of ANKRD11. ANK: ankyrin repeat domain, RD1: repression domain 1, AD: activation domain, RD2: repression domain 2
Hotspot variants of ANKRD11 protein
Mutation rates vary significantly along nucleotide sequences such that variants often concentrate at certain positions called hotspots [ 100 ]. DNA sequences prone to variation are highly dependent of gene sequence and structure as well as its chromosomal location, such as GC-rich region, microsatellites, meiotic recombination, nonallelic homologous recombination, centromeric rearrangements, telomeres and subtelomeric regions, replication timing and common fragile sites [ 101 , 102 ]. Therefore, hotspot variants are indicative of the structural and functional properties of DNA sequence. Within the spectrum of ANKRD11 variants, over two dozen distinct variants have been identified in at least three patients. Beyond a few variants that have been vertically inherited within a single family, the majority of variants were discovered in multiple sporadic patients, underscoring the propensity for these genetic variants to arise independently in unrelated individuals. Four hotspot variants of ANKRD11 protein were observed including p.Glu461Glnfs*48, p.Lys635Glnfs*26, p.Glu800Asnfs*62 and p.Lys803Argfs*5 (Fig. 3 A). These four variants are frameshift variants generated by c.1381_1384delGAAA, c.1903_1907delAAACA, c.2395_2398delAAAG and c.2408_2412delAAAAA, respectively. Two additional prevalent frameshift variants were traced back to analogous genomic alterations including p.Asn725Lysfs*23 and p.Thr462Lysfs*47 arising from c.2175_2178delCAAA and c.1385_1388delCAAA, respectively. The propensity for short deletions within AAA-type-containing sequences may be associated with polymerase slippage events induced by tandem repeats, a well-established mechanism for indels [ 100 ]. Nonetheless, it should be highlighted that CCC-type-containing sequences exhibit a heightened vulnerability to this form of genetic variation [ 103 , 104 ]. RD2 domain located at the C-terminus of ANKRD11 seemed to be particularly vulnerable to a range of variant events in KBGS patients, with missense variants being notably prevalent (Fig. 3 A). Conversely, the missense variants occuring in RD2 domain were relatively rare in general population (Fig. 3 B). This was consistent with the results of in vitro cellular assays, which showed that missense variants occurring in the RD2 domain impaired the protein function of ANKRD11 [ 66 ]. Some frameshift and nonsense variants of ANKRD11 have been identified in general population, such as p.Glu2082Argfs*20, p.Ser2180Phefs*6, p.Glu1075* and p.Gln2507*, indicating a pattern of variable expressivity and incomplete penetrance associated with ANKRD11 variants [ 2 ]. Taken together, the presence of hotspot variants offers valuable insights into the inherent vulnerability of specific DNA sequence to abnormal DNA repair, replication, and modification or environmental exposures. These findings warrant in-depth exploration at the molecular level to unravel the underlying mechanisms and implications.

Frequency of ANKRD11 variants in a total of 838 KBGS patients ( A ) and ANKRD11 allele frequency in general population ( B ). ANKRD11 allele frequency below 1% in general poulation was obtained from gnomAD ( http://gnomad-sg.org/ ). The abscissa represents the full-length amino acid sequence of ANKRD11, and the ordinate represents the frequency
ANKRD11 variants and short stature in patients with KBGS
Frequency of occurrence of short stature in patients with ankrd11 variants.
Short stature is defined as height less than − 2 standard deviation (SD) or below the third percentile of corresponding mean height for age-, gender- and race-matched populations [ 105 , 106 ]. As widely recognized, height is a highly heritable characteristic, and is classically influenced by hundreds of common variants pinpointed by genome-wide association studies (GWAS) [ 107 , 108 ]. By comparison, the impact of rare and low-frequency monogenic variants on height is more pronounced, yielding a larger effect size compared to single nucleotide polymorphisms (SNPs) [ 109 , 110 ]. Finding new genes with rare deleterious variants relating to growth is of considerable significance. Case series and individual reports serve as valuable sources of evidence for investigating the frequency of occurrence of short stature among patients harboring ANKRD11 variants. In 121 patients reported with height SDS, a significant proportion, amounting to 48.76% (59/121), exhibited a height below the − 2 SDS (Fig. 4 A). This prevalence was observed with nearly equal frequency across genders, with female patients exhibiting a rate of 46.43% (26/56) and male patients exhibiting a rate of 49.02% (25/51). The height SDS of females and males were − 1.80 ± 1.27 and − 1.85 ± 1.28 SDS, respectively. Upon incorporating additional patients recorded with height percentile values into the analysis, the proportion of patients with short stature was found to be 47.35% (116/245). Moreover, while some patients did not exhibit short stature, their adult height SDS or growth percentile might be lower than expected if their genetic potential (mid-parental height) was taken into account. However, most studies did not report patients’ genetic potential for height, making it challenging to extract this specific information from the published literature. Overall, approximately half of the patients with ANKRD11 variants exhibited short stature, consequently, this characteristic stand as an important manifestation of KBGS attributable to ANKRD11 variants. Certainly, compared to other features, the incidence of short stature was less frequent than that of craniofacial anomalies (100%), dental anomalies (80%) and intellectual disability (77%) [ 48 ]. Notably, patients with ANKRD11 variants displayed a variable height phenotype ranging from as low as -4.9 SDS to as high as + 1.5 SDS. It can be ascribed to several factors, including genetic context of the gene, modified penetrance, variant type and variant location [ 111 , 112 ]. There was no significant difference in height SDS among patients with ANKRD11 variants located in different regions or with different ANKRD11 variant types ( p > 0.05) (Fig. 4 B&C). Previous investigation has revealed that terminations close to the C-terminus of the ANKRD11 protein tended to have less severe short stature, but the research did not yield a statistically significant difference or a clear trend in the severity of short stature among the various types of ANKRD11 variants [ 39 ]. The findings of the current study indicated that no genotype-phenotype correlation was established. Certainly, a limited number of patients with ANKRD11 variants across different domains present a significant constraint on this conclusion.

Distribution of gender and height SDS of patients having ANKRD11 variants ( A ) and comparison of height SDS of patients having ANKRD11 variants within different domain ( B ) or having different ANKRD11 variant types ( C ). ANK: ankyrin repeat domain, RD1: repression domain 1, AD: activation domain, RD2: repression domain 2
Frequency of ANKRD11 pathogenic variants in short-stature cohorts
Functional variants in the ANKRD11 gene have been identified through exome sequencing or gene panels in multiple short-stature cohorts (Table 1 ). The frequency of pathogenic variants was estimated to be between 0.35% and 0.55% [ 43 , 68 , 79 , 113 ]. These variants were identified in patients initially diagnosed as having syndromic short stature, however, subsequent molecular diagnosis facilitated a more precise diagnosis of KBG syndrome. Syndromic short stature represents a phenotypic and genetically heterogeneous disease, and it accounts for a large part of the etiology of short stature. Considering the wide range of phenotypic manifestations and variable degree of severity, certain patients with short stature suffering from KBGS may not be accurately diagnosed in clinical practice. Consequently, it is likely that these patients harbor rare pathogenic variants in the ANKRD11 gene, which may elude detection and result in their classification within the vast and enigmatic group of short stature with undetermined etiologies. Genetic testing should be factored into precise diagnosis of syndromic short stature in the future. Based on previous studies estimating the occurrence of short stature at approximately 3% [ 114 , 115 , 116 ], the prevalence of ANKRD11 variants in the general population could be roughly calculated to be in the range of 0.0105–0.0165%. Nevertheless, given the limited sample sizes and the variability among different cohorts studied for short stature, the frequency of ANKRD11 variants remains uncertain and requires a more accurate assessment. This evaluation should ideally be conducted through large-scale population screenings, employing artificial intelligence-enhanced phenotyping in conjunction with genetic testing [ 117 ]. Despite the growing awareness and attention this condition has recently garnered in the clinical and genetic research communities, there remains a significant gap in the identification and management of KBGS patients. Therefore, the development of international consensus guidelines for the diagnosis of KBGS is of paramount importance.
Recombinant human growth hormone therapy
In 1985, recombinant human growth hormone (rhGH) received approval from the US Food and Drug Administration (FDA) for the treatment of children with severe GHD. Since then, over the past nearly forty years, the application of rhGH has been progressively expanded to enhance the height outcomes in children with a variety of growth disorders, including chronic renal insufficiency (CRI), ISS, SGA without catch-up growth, Prader-Willi Syndrome (PWS), Noonan syndrome (NS), Turner syndrome (TS) and SHOX haploinsufficiency [ 118 , 119 ]. The advent of high-throughput sequencing technology has ushered in a period of rapid advancement in the field of genetics and genomics, and this progress has significantly broadened our capacity for diagnosing and treating conditions associated with short stature. We are now entering a transformative era characterized by molecular diagnosis and the tailoring of therapeutic interventions to the specific genetic makeup of individuals, including their responsiveness to rhGH therapy [ 120 ]. It has been observed that pathogenic variants in the aggrecan ( ACAN ), natriuretic peptide receptor 2 ( NPR2 ), and Indian hedgehog ( IHH ) genes, which are integral to growth plate development, have been consistently associated with a positive response to rhGH therapy [ 121 , 122 , 123 , 124 , 125 ]. In this review, we delineated the growth response observed in patients harboring ANKRD11 variants who received rhGH therapy (Table 2 ). The ages at initiation of rhGH treatment ranged from 5.2 to 14 years, and the treatment duration extended from 0.58 to 3 years. Following rhGH treatment, all patients exhibited varying levels of catch-up growth, as reflected by a range in Δ height SDS from 0.14 to 1.87. Among the nine patients, five showed a significant height improvement, reaching values above − 2 SDS ( -0.75 SDS for patient 3, − 0.7 SDS for patient 4, -1.86 SDS for patient 5, -1.8 SDS for patient 8 and − 1.91 SDS for patient 9). Most patients displayed either a good or moderate response to rhGH therapy. However, there was an exception with patient 3, a 7.9-year-old girl, whose height SDS only increased by 0.14 following a continuous treatment period of 0.58 years. Practically, a four-year-old girl form Australia with ANKRD11 variant (c.6472G > T, p.Glu2158*), showed no response to rhGH therapy [ 49 ]. The girl was not included in Table 2 due to the lack of height data. The potential existence of additional factors that may be contributing to the suboptimal response to rhGH remains uncertain.
Given the evidence suggesting that the ANKRD11 gene acts as a potential tumor suppressor due to its interaction with the p53 protein, particular attention should be paid to the safety profile of rhGH therapy, particularly oncogenic risks [ 126 ]. However, observational studies have reported no increased risk of mortality or the development of primary cancers among pediatric patients receiving rhGH treatment [ 127 , 128 , 129 ]. The implementation of cancer surveillance in patients clinically diagnosed as having KBGS due to ANKRD11 variants has been previously contemplated, and few patients were reported to develop malignant tumors [ 130 , 131 ]. Short stature is one of all KBGS phenotypes that can be effectively treated with growth-promoting drugs, but there are few patients receiving rhGH treatment. The approval and accessibility of rhGH therapy for KBGS may be limited in certain countries, which highlights the imperative for further investigation and research within this specialized domain. In alignment with the recommendations proposed by Reynaert et al. [ 58 ], we advocate for a more favorable stance towards the implementation of short-term rhGH therapy for ANKRD11 variant-induced KBGS patients with severe short stature.
Underlying mechanisms of ANKRD11 variants causing short stature
Human longitudinal bone growth is persistently driven by the process of endochondral ossification within the epiphyseal growth plate that is characterized by three histologically distinct zones (resting, proliferative, and hypertrophic zones) throughout the stages of postnatal development [ 132 ]. As the slowly-cycling reserve cells, resting chondrocytes are maintained in a wingless-related integration site (Wnt)-inhibitory environment, and it contains a certain proportion of parathyroid hormone-related protein (PTHrP)-expressing skeletal stem-like cells producing rapidly proliferating columnar chondrocytes parallel to the direction of bone elongation [ 133 ]. Proliferative zone chondrocytes will differentiate into hypertrophic chondrocytes characterized by specific expression of type X collagen gene ( Col10a1 ), and further undergo apoptosis or osteoblasts trans-differentiation, thereby contributing to bone elongation [ 134 , 135 ]. The orchestrated differentiation of chondrocytes within the growth plate is governed by a complex interplay of numerous genes that are involved in a variety of signaling pathways, including hormonal signaling, paracrine signaling, intracellular pathways and extracellular matrix homeostasis (Fig. 5 ) [ 68 , 136 , 137 , 138 ]. Functional variants in any of these genes can disrupt the growth plate chondrogenesis and impair the subsequent bone elongation. It was hypothesized that ANKRD11 plays a direct role in the transcriptional regulation of certain critical genes via intracellular pathways in the process of growth plate development [ 68 ]. In a prior investigation, Yoda mice with an N-ethyl-N-nitrosourea (ENU)-induced mutation in the ANKRD11 gene, exhibited a markedly reduced body size and presented with a phenotype reminiscent of osteoporosis compared to littermate controls [ 3 ]. However, no alterations were observed in the histological structure of the tibial growth plate and plasma IGF-1 level between six-month-old Yoda mice and wild-type mice. Given that growth plate in rodents do not undergo fusion but are instead subject to an age-related decrease following sexual maturation [ 139 ], it can be inferred that adult mice with ANKRD11 deficiency may not well accurately reflect the aberrant differentiation process of growth plate chondrocytes during rapid bone elongation. Data obtained from the International Mouse Phenotyping Consortium (IMPC) indicate that C57BL/6 N mice carrying a heterozygous ANKRD11 tm1b(EUCOMM)Wtsi allele exhibited a reduction in body length when compared to their littermate controls ( https://www.mousephenotype.org/data/genes/MGI:1924337 ). Additionally, mice with a conditional deletion of the ANKRD11 gene in neural crest cells dispalyed ossification centers that were either incapable of expansion or failed to fuse, demonstrating the critical regulatory role of ANKRD11 gene in intramembranous ossification [ 22 ]. In vitro studies further revealed that ANKRD11 was capable of enhancing the transactivation of the p21 gene, a key factor in the chondrogenic differentiation of ATDC5 cells induced by insulin supplements [ 61 ]. The chondrogenic differentiation of ATDC5 cells induced by insulin-transferrin-selenium is a widely recognized in vitro model mimicking endochondral ossification [ 140 , 141 , 142 , 143 ]. The potential role of the ANKRD11-p21 signaling pathway in growth plate development as a plausible mechanism to elucidate the short stature observed in KBGS patients warrants further investigation. To elucidate the functional mechanisms of the ANKRD11 gene in the physiological process of growth plate development, it is essential to conduct further study employing a mouse model with chondrocyte-specific ANKRD11 ablation, utilizing the CRISPR/Cas9 and Cre/LoxP recombination system.

Disease-causing genes associated with short stature through affecting the endochondral ossification of epiphyseal growth plate. The ANKRD11 gene may be implicated in this process as a transcription regulator. RZ: resting zone, PZ: proliferative zone, PHZ: prehypertrophic zone, HZ: hypertrophic zone
Conclusions
Frameshift and nonsense were the most common types of ANKRD11 variants. Approximately half of the KBGS patients harboring ANKRD11 variants had short stature. However, the current study has not established a clear correlation between the genotype and this phenotypic manifestation. Some patients harboring ANKRD11 variants may initially be diagnosed as syndromic short stature due to limited recognition of KBGS. While patients with ANKRD11 variants exhibit a positive response to rhGH therapy, further investigation is warranted to substantiate its efficacy and safety. Functional variants in the ANKRD11 gene can potentially disrupt the longitudinal growth of bones by influencing the orderly differentiation process of growth plate chondrocytes, which needs deeper investigation through fundamental research to elucidate its underlying mechanisms.
Data availability
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
Abbreviations
American college of medical genetics and genomics
Activation domain
Alteration/deficiency in activation 3
Amplified in breast cancer 1
Association for molecular pathology
Ankyrin repeats-containing cofactor
Ankyrin repeat domain containing-protein 11
One-way analysis of variance
Brain-derived neurotrophic factor
[(CAMP-response-element binding protein)-binding protein]-associated factor
(CAMP-response-element binding protein)-binding protein
CAMP-response-element binding protein
Chronic renal insufficiency
Electroencephalogram
N-ethyl-N-nitrosourea
Estrogen receptor
Food and drug administration
Growth hormone
Growth hormone deficiency
Growth hormone insensitivity
Genome-wide association study
Histone acetylase
Histone deacetylase
Histone 3 lysine 9
Histone 4 lysine 5
Histone 4 lysine 8
Histone 4 lysine 16
Height standard deviation score
Hypertrophic zone
Insulin-like growth factor
Insulin-like growth factor binding protein 3
Isolated growth hormone deficiency
Indian hedgehog
International mouse phenotyping onsortium
Insertion or deletion
Intelligence quotient
Idiopathic short stature
Ligand-binding domain
Multiple pituitary hormone deficiency
Magnetic resonance imaging
Nuclear localization signal
Natriuretic peptide receptor 2
Nuclear receptors
Noonan syndrome
Online mendelian inheritance in man
Per-Arnt-Sim
Parathyroid hormone-related protein
Protein-truncating variant
Prader-Willi syndrome
Proliferative zone
Repression domain
Recombinant human growth hormone
Resting zone
Standard deviation
Standard deviation score
Small for gestational age
Single nucleotide polymorphism
Secondary ossification center
Transcriptional enhanced associate domain
Tyrosine receptor kinase B
Turner syndrome
Whole exome sequencing
Wingless-related integration site
Yes-associated protein
Kang Y, He D, Li Y, Zhang Y, Shao Q, Zhang M, et al. A heterozygous point mutation of the ANKRD11 (c.2579C > T) in a Chinese patient with idiopathic short stature. Mol Genet Genomic Med. 2019;7(12):e988.
Article CAS PubMed PubMed Central Google Scholar
Parenti I, Mallozzi MB, Huning I, Gervasini C, Kuechler A, Agolini E, et al. ANKRD11 variants: KBG syndrome and beyond. Clin Genet. 2021;100(2):187–200.
Article CAS PubMed Google Scholar
Barbaric I, Perry MJ, Dear TN, Rodrigues Da Costa A, Salopek D, Marusic A, et al. An ENU-induced mutation in the Ankrd11 gene results in an osteopenia-like phenotype in the mouse mutant Yoda. Physiol Genomics. 2008;32(3):311–21.
Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13(6):1435–48.
Zhang AH, Li CW, Chen JD. Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem Bioph Res Co. 2007;358(4):1034–40.
Article CAS Google Scholar
Neilsen PM, Cheney KM, Li CW, Chen JD, Cawrse JE, Schulz RB, et al. Identification of ANKRD11 as a p53 coactivator. J Cell Sci. 2008;121(21):3541–52.
Zhang AH, Yeung PL, Li CW, Tsai SC, Dinh GK, Wu XY, et al. Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J Biol Chem. 2004;279(32):33799–805.
Li CW, Dinh GK, Zhang AH, Chen JD. Ankyrin repeats-containing cofactors interact with ADA3 and modulate its co-activator function. Biochem J. 2008;413:349–57.
Whitmore SA, Crawford J, Apostolou S, Eyre H, Baker E, Lower KM, et al. Construction of a high-resolution physical and transcription map of chromosome 16q24.3: a region of frequent loss of heterozygosity in sporadic breast cancer. Genomics. 1998;50(1):1–8.
Powell JA, Gardner AE, Bais AJ, Hinze SJ, Baker E, Whitmore S, et al. Sequencing, transcript identification, and quantitative gene expression profiling in the breast cancer loss of heterozygosity region 16q24.3 reveal three potential tumor-suppressor genes. Genomics. 2002;80(3):303–10.
Garee JP, Chien CD, Li JV, Wellstein A, Riegel AT. Regulation of HER2 oncogene transcription by a multifunctional coactivator/corepressor complex. Mol Endocrinol. 2014;28(6):846–59.
Article PubMed PubMed Central Google Scholar
Kushner MH, Ory V, Graham GT, Sharif GM, Kietzman WB, Thevissen S, et al. Loss of ANCO1 repression at AIB1/YAP targets drives breast cancer progression. Embo Rep. 2020;21(1):e48741.
Lim SP, Wong NC, Suetani RJ, Ho K, Ng JL, Neilsen PM, et al. Specific-site methylation of tumor suppressor ANKRD11 in breast cancer. Eur J Cancer. 2012;48(17):3300–9.
Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975;11(5):7–18.
CAS PubMed Google Scholar
Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89(2):289–94.
Morel Swols D, Foster IIJ, Tekin M. KBG syndrome. Orphanet J Rare Dis. 2017;12(1):183.
Cucco F, Sarogni P, Rossato S, Alpa M, Patimo A, Latorre A, et al. Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia De Lange syndrome. Am J Med Genet A. 2020;182(7):1690–6.
Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M, et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and coffin-siris-like syndrome. J Hum Genet. 2017;62(8):741–6.
Walz K, Cohen D, Neilsen PM, Foster IIJ, Brancati F, Demir K, et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum Genet. 2015;134(2):181–90.
Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, et al. Ankrd11 is a chromatin Regulator involved in Autism that is essential for neural development. Dev Cell. 2015;32(1):31–42.
Ka MH, Kim WY. ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol Dis. 2018;111:138–52.
Roth DM, Baddam P, Lin HM, Vidal-Garcia M, Aponte JD, De Souza ST, et al. The chromatin regulator Ankrd11 controls palate and cranial bone development. Front Cell Dev Biol. 2021;9:645836.
Article Google Scholar
Yana K, Elia A, Ronan N, Adrianne W, Irina P, Nicole D, et al. The chromatin regulator Ankrd11 controls cardiac neural crest cell-mediated outflow tract remodeling and heart function. Nat Commun. 2024;15:4632.
Elena MC, Fiona BK, Fermina LG, Saoud TS, Rosario LR, Rebeca LDP, Ignacio MF, Beatriz M, et al. Clinical description, molecular delineation and genotype-phenotype correlation in 340 patients with KBG syndrome: addition of 67 new patients. J Med Genet. 2023;60:644–54.
Xu MZ, Zhou HL, Yong J, Cong PK, Li CJ, Yu YS, et al. A Chinese patient with KBG syndrome and a 9q31.2-33.1 microdeletion. Eur J Med Genet. 2013;56(5):245–50.
Article PubMed Google Scholar
Benson KA, White M, Allen NM, Byrne S, Carton R, Comerford E, et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet. 2020;28(8):1066–77.
Chen J, Xia ZM, Zhou YL, Ma XM, Wang XD, Guo QW. A de novo frameshift variant of ANKRD11 (c.1366_1367dup) in a Chinese patient with KBG syndrome. BMC Med Genomics. 2021;14(1):68.
Kim HJ, Cho E, Park JB, Im WY, Kim HJ. A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur J Med Genet. 2015;58(2):86–94.
Low K, Hills A, Williams M, Duff-Farrier C, McKee S, Smithson SF. A splice-site variant in ANKRD11 associated with classical KBG syndrome. Am J Med Genet A. 2017;173(10):2844–6.
Tanaka Y, Morisada N, Suzuki T, Ohashi Y, Ye MJ, Nozu K, et al. A woman with a dual genetic diagnosis of autosomal dominant tubulointerstitial kidney disease and KBG syndrome. CEN Case Rep. 2021;10(2):184–8.
de la Jimenez M, Fernandez-Mayoralas DM, Lopez-Martin S, Albert J, Calleja-Perez B, Fernandez-Perrone AL, et al. Abnormal frontal gyrification pattern and uncinate development in patients with KBG syndrome caused by ANKRD11 aberrations. Eur J Paediatr Neurol. 2021;35:8–15.
Cianci P, Pezzoli L, Maitz S, Agosti M, Iascone M, Selicorni A. Dual genetic diagnoses: neurofibromatosis type 1 and KBG syndrome. Clin Dysmorphol. 2020;29(2):101–3.
Li Q, Yang L, Wu J, Lu W, Zhang M, Luo F. A case of KBG syndrome caused by mutation of ANKRD11 gene and literature review. Chin J Evid Based Pediatr. 2018;13(6):452–8.
Google Scholar
Bianchi PM, Bianchi A, Digilio MC, Tucci FM, Sitzia E, De Vincentiis GC. Audiological findings in a de novo mutation of ANKRD11 gene in KBG syndrome: report of a case and review of the literature. Int J Pediatr Otorhinolaryngol. 2017;103:109–12.
Parenti I, Gervasini C, Pozojevic J, Graul-Neumann L, Azzollini J, Braunholz D, et al. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia De Lange-overlapping phenotype. Clin Genet. 2016;89(1):74–81.
Wojciechowska K, Nurzynska-Flak J, Styka B, Kacprzak M, Lejman M. Case report: two newly diagnosed patients with KBG syndrome-two different molecular changes. Front Pediatr. 2021;9:649043.
Low K, Ashraf T, Canham N, Clayton-Smith J, Deshpande C, Donaldson A, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016;170(11):2835–46.
Goldenberg A, Riccardi F, Tessier A, Pfundt R, Busa T, Cacciagli P, et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. 2016;170(11):2847–59.
Li Q, Sun C, Yang L, Lu W, Luo F. Comprehensive analysis of clinical spectrum and genotype associations in Chinese and literature reported KBG syndrome. Transl Pediatr. 2021;10(4):834–42.
Aoi H, Mizuguchi T, Ceroni JR, Kim VEH, Furquim I, Honjo RS, et al. Comprehensive genetic analysis of 57 families with clinically suspected Cornelia De Lange syndrome. J Hum Genet. 2019;64(10):967–78.
Mattei D, Cavarzere P, Gaudino R, Antoniazzi F, Piacentini G. DYSMORPHIC features and adult short stature: possible clinical markers of KBG syndrome. Ital J Pediatr. 2021;47(1):15.
Ockeloen CW, Willemsen MH, de Munnik S, van Bon BWM, de Leeuw N, Verrips A, et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet. 2015;23(9):1176–85.
Homma TK, Freire BL, Kawahira RSH, Dauber A, Funari MFD, Lerario AM, et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J Pediatr. 2019;215:192–8.
Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, et al. Genetic heterogeneity in Cornelia De Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51(10):659–68.
Wong JKL, Campbell D, Ngo ND, Yeung F, Cheng G, Tang CSM, et al. Genetic study of congenital bile-duct dilatation identifies de novo and inherited variants in functionally related genes. BMC Med Genomics. 2016;9:75.
Ge XY, Ge L, Hu WW, Li XL, Hu YY. Growth hormone therapy for children with KBG syndrome: a case report and review of literature. World J Clin Cases. 2020;8(6):1172–9.
Alfieri P, Caciolo C, Lazzaro G, Menghini D, Cumbo F, Dentici ML, et al. Cognitive and adaptive characterization of children and adolescents with KBG syndrome: an explorative study. J Clin Med. 2021;10(7):1523.
Gao F, Zhao X, Cao B, Fan X, Li X, Li L, et al. Genetic and phenotypic spectrum of KBG syndrome: a report of 13 new Chinese cases and a review of the literature. J Pers Med. 2022;12(3):407.
Murray N, Burgess B, Hay R, Colley A, Rajagopalan S, McGaughran J, et al. KBG syndrome: an Australian experience. Am J Med Genet A. 2017;173(7):1866–77.
Gnazzo M, Lepri FR, Dentici ML, Capolino R, Pisaneschi E, Agolini E, et al. KBG syndrome: common and uncommon clinical features based on 31 new patients. Am J Med Genet A. 2020;182(5):1073–83.
Sayed ISM, Abdel-Hamid MS, Abdel-Salam GMH. KBG syndrome in two patients from Egypt. Am J Med Genet A. 2020;182(6):1309–12.
Kleyner R, Malcolmson J, Tegay D, Ward K, Maughan A, Maughan G, et al. KBG syndrome involving a single-nucleotide duplication in ANKRD11. Cold Spring Harb Mol Case Stud. 2016;2(6):a001131.
Libianto R, Wu KH, Devery S, Eisman JA, Center JR. KBG syndrome presenting with brachydactyly type E. Bone. 2019;123:18–22.
Alves RM, Uva P, Veiga MF, Oppo M, Zschaber FCR, Porcu G, et al. Novel ANKRD11 gene mutation in an individual with a mild phenotype of KBG syndrome associated to a GEFS + phenotypic spectrum: a case report. BMC Med Genet. 2019;20(1):16.
Scarano E, Tassone M, Graziano C, Gibertoni D, Tamburrino F, Perri A, et al. Novel mutations and unreported clinical features in KBG syndrome. Mol Syndromol. 2019;10(3):130–8.
Alfieri P, Demaria F, Licchelli S, Santonastaso O, Caciolo C, Digilio MC et al. Obsessive compulsive symptoms and psychopathological profile in children and adolescents with KBG syndrome. Brain Sci. 2019;9(11).
De Bernardi ML, Ivanovski I, Caraffi SG, Maini I, Street ME, Bayat A, et al. Prominent and elongated coccyx, a new manifestation of KBG syndrome associated with novel mutation in ANKRD11. Am J Med Genet A. 2018;176(9):1991–5.
Reynaert N, Ockeloen CW, Savendahl L, Beckers D, Devriendt K, Kleefstra T, et al. Short stature in KBG syndrome: first responses to growth hormone treatment. Horm Res Paediatr. 2015;83(5):361–4.
Bayat A, Møller LB, Hjortshøj TD. Første Danske patient med et genkendeligt genetisk KBG-syndrom. Ugeskr Læger. 2018;180:V11170848.
PubMed Google Scholar
Latorre-Pellicer A, Ascaso Á, Lucia-Campos C, Gil-Salvador M, Arnedo M, Antoñanzas R, et al. Things are not always what they seem: from Cornelia De Lange to KBG phenotype in a girl with genetic variants in NIPBL and ANKRD11. Mol Genet Genomic Med. 2021;9(11):e1826.
Zhang TT, Yang Y, Yin XL, Wang XQ, Ni JH, Dong ZY, et al. Two loss-of-function ANKRD11 variants in Chinese patients with short stature and a possible molecular pathway. Am J Med Genet A. 2021;185(3):710–8.
Kim SJ, Yang A, Park JS, Kwon DG, Lee JS, Kwon YS, et al. Two novel mutations of ANKRD11 gene and wide clinical spectrum in KBG syndrome: case reports and literature review. Front Genet. 2020;11:579805.
Butler MG, Rafi SK, Hossain W, Stephan DA, Manzardo AM. Whole exome sequencing in females with autism implicates novel and candidate genes. Int J Mol Sci. 2015;16(1):1312–35.
Abe-Hatano C, Iida A, Kosugi S, Momozawa Y, Terao C, Ishikawa K, et al. Whole genome sequencing of 45 Japanese patients with intellectual disability. Am J Med Genet A. 2021;185(5):1468–80.
Kutkowska-Kazmierczak A, Boczar M, Kalka E, Castaneda J, Klapecki J, Pietrzyk A, et al. Wide fontanels, delayed speech development and hoarse voice as useful signs in the diagnosis of KBG syndrome: a clinical description of 23 cases with pathogenic variants involving the ANKRD11 gene or submicroscopic chromosomal rearrangements of 16q24.3. Genes-Basel. 2021;12(8):1257.
de Boer E, Ockeloen CW, Kampen RA, Hampstead JE, Dingemans AJM, Rots D, et al. Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein. Genet Med. 2022;24(10):2051–64.
Hong JH, Kim SH, Lee ST, Choi JR, Kang HC, Lee JS, et al. Early diagnosis of KBG syndrome using diagnostic exome sequencing. J Korean Child Neurol Soc. 2018;26(4):272–75.
Li X, Yao R, Chang GY, Li Q, Song C, Li N, et al. Clinical profiles and genetic spectra of 814 Chinese children with short stature. J Clin Endocr Metab. 2022;107(4):972–85.
Nardello R, Mangano GD, Antona V, Fontana A, Striano P, Giorgio E, et al. Electroclinical features and outcome of ANKRD11-related KBG syndrome: a novel report and literature review. Seizure. 2021;85:151–4.
Samanta D, Willis E. Electroencephalographic findings in KBG syndrome: a child with novel mutation in ANKRD11 gene. Acta Neurol Belg. 2015;115(4):779–82.
Popp B, Ekici AB, Thiel CT, Hoyer J, Wiesener A, Kraus C, et al. Exome Pool-Seq in neurodevelopmental disorders. Eur J Hum Genet. 2017;25(12):1364–76.
Aitken S, Firth HV, McRae J, Halachev M, Kini U, Parker MJ, et al. Finding diagnostically useful patterns in quantitative phenotypic data. Am J Hum Genet. 2019;105(5):933–46.
Miao P, Feng JH, Guo YF, Wang JD, Xu XX, Wang Y, et al. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin Genet. 2018;94(6):512–20.
Meyer R, Soellner L, Begemann M, Dicks S, Fekete G, Rahner N, et al. Targeted next generation sequencing approach in patients referred for silver-Russell syndrome testing increases the mutation detection rate and provides decisive information for clinical management. J Pediatr. 2017;187:206–12.
Reuter MS, Chaturvedi RR, Liston E, Manshaei R, Aul RB, Bowdin S, et al. The cardiac genome clinic: implementing genome sequencing in pediatric heart disease. Genet Med. 2020;22(6):1015–24.
Cao YH, Zhang LY, Cao KF, Zhang GY. KBG syndrome: a case report and literature review. J Clin Pediatr. 2020;38(5):335–38.
Yang YY, Wen PQ, Su Z, Wang L, Zhao X. Gender difference in clinical manifestations of KBG syndrome due to variants of ANKRD11 gene. Chin J Med Genet. 2021;7:663–66.
Wang DY, Lai PJ, Li XB. Analysis of ANKRD11 gene variant in a family affected with KBG syndrome. Chin J Med Genet. 2020;37(9):1029–31.
Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet Med. 2018;20(6):630–8.
Bestetti I, Crippa M, Sironi A, Tumiatti F, Masciadri M, Smeland MF, et al. Expanding the molecular spectrum of ANKRD11 gene defects in 33 patients with a clinical presentation of KBG syndrome. Int J Mol Sci. 2022;23(11):5912.
Wei SS, Li YY, Yang WL, Chen SX, Liu FP, Zhang M, et al. Functional investigation of a novel ANKRD11 frameshift variant identified in a Chinese family with KBG syndrome. Heliyon. 2024;10(6):e28082.
Luca M, Elena C, Fjorilda C, Alice S, Roberto C, Sandra D, et al. A case of early-onset Parkinson’s disease in a patient with KBG syndrome. Neurol Sci. 2023;44:4537–39.
Shangguan HK, Wang J, Lin JD, Huang XZ, Zeng Y, Chen RM. A study on genotypes and phenotypes of short stature caused by epigenetic modification gene variants. Eur J Pediatr. 2024;183(3):1403–14.
Zain A, Sanaa C, Cheryl C, Fowzan A, Stephen S, Sofia F, et al. ANKRD11 pathogenic variants and 16q24.3 microdeletions share an altered DNA methylation signature in patients with KBG syndrome. Hum Mol Genet. 2023;32(9):1429–38.
Adelaide C, Camilla M, Ludovica P, Davide P, Fulvio DA, Veronica CB, et al. Cerebellar heterotopia: broadening the neuroradiological spectrum of KBG syndrome. Cerebellum. 2024;23:1736–40.
Zhang HZ, Guo XN, Yang C, Zhang KH, Wang D, Wang J, et al. Clinical feature and genetic mutation of KBG syndrome diagnosed in neonatal period: a case report. Medicine. 2023;102(40):e35449.
Francesca P, Stefano GC, Gianluca C, Lara V, Manuela N, Giorgia C, et al. Deep phenotyping of the neuroimaging and skeletal features in KBG syndrome: a study of 53 patients and review of the literature. J Med Genet. 2023;60(12):1224–34.
Ola K, Kathleen S, Drake C, Lily G, Elaine M, Anastassia V, et al. Documentation and prevalence of prenatal and neonatal outcomes in a cohort of individuals with KBG syndrome. Am J Med Genet A. 2023;191(9):2364–75.
Robyn W, Madeline K, Sangeetha Y, Gregory C, Puneet J. Epilepsy in KBG Syndrome: report of additional cases. Pediatr Neurol. 2024;151:138–42.
Auconi M, Serino D, Digilio MC, Gnazzo M, Conti M, Vigevano F, et al. Epilepsy in KBG syndrome. Dev Med Child Neurol. 2023;65(5):712–20.
Eoin PD, Kathleen MG, Amre S, Nicholas MA, et al. Epileptic dyskinetic encephalopathy in KBG syndrome: expansion of the phenotype. Epilepsy Behav Rep. 2024;25:100647.
Anna A, Lior G, Shahar St, Gundula P, Ayan M, Zhong R, et al. Genetic insights into childhood-onset schizophrenia: the yield of clinical exome sequencing. Schizophr Res. 2023;252:138–45.
Nada A, Siham CE, Maria Z, Amal C, Lamia A, Amal TI, et al. Identification of two novel ANKRD11 mutations: highlighting incomplete penetrance in KBG syndrome. Ann Lab Med. 2024;44(1):110–7.
Choi YH, Choi JM, Do HS, Hwang SJ, Seo GH, Choi IH, et al. KBG syndrome: clinical features and molecular findings in seven unrelated Korean families with a review of the literature. Mol Genet Genomic Med. 2023;11(4):e2127.
Miyake N, Tsurusaki Y, Fukai R, Kushima I, Okamoto N, Ohashi K, et al. Molecular diagnosis of 405 individuals with autism spectrum disorder. Eur J Hum Genet. 2023. https://doi.org/10.1038/s41431-023-01335-7 .
Wang L, Li J, Xu J, Xu Y, Wang J, Feng Y, et al. Clinical and genetic analysis of three children with KBG syndrome due to novel variants of ANKRD11 gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2023;40(1):1–6.
Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186–93.
Sevim Bayrak C, Stein D, Jain A, Chaudhary K, Nadkarni GN, Van Vleck TT, et al. Identification of discriminative gene-level and protein-level features associated with pathogenic gain-of-function and loss-of-function variants. Am J Hum Genet. 2021;108(12):2301–18.
Fu JM, Satterstrom FK, Peng MS, Brand H, Collins RL, Dong S, et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet. 2022;54(9):1320–31.
Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res-Rev Mutat. 2003;544(1):65–85.
Nesta AV, Tafur D, Beck CR. Hotspots of human mutation. Trends Genet. 2021;37(8):717–29.
Seplyarskiy VB, Sunyaev S. The origin of human mutation in light of genomic data. Nat Rev Genet. 2021;22(10):672–86.
Taylor MS, Ponting CP, Copley RR. Occurrence and consequences of coding sequence insertions and deletions in mammalian genomes. Genome Res. 2004;14(4):555–66.
Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZDD, Mu XJ, et al. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 2013;23(5):749–61.
Wang PP, Ji BL, Shao Q, Zhang M, Ban B. Association between insulin-like growth factor-1 and uric acid in Chinese children and adolescents with idiopathic short stature: a cross-sectional study. Biomed Res Int. 2018;2018:4259098.
PubMed PubMed Central Google Scholar
Zhao Q, Zhang M, Chu Y, Sun H, Pan H, Ban B. A retrospective analysis of patients with short stature in Eastern China between 2013 and 2019. Biomed Res Int. 2021;2021:6640026.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–9.
Durand C, Rappold GA. Height matters-from monogenic disorders to normal variation. Nat Rev Endocrinol. 2013;9(3):171–7.
Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. 2017;542(7640):186–90.
Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99(9):3080–92.
Vu V, Verster AJ, Schertzberg M, Chuluunbaatar T, Spensley M, Pajkic D, et al. Natural variation in gene expression modulates the severity of mutant phenotypes. Cell. 2015;162(2):391–402.
Castel SE, Cervera A, Mohammadi P, Aguet F, Reverter F, Wolman A, et al. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat Genet. 2018;50(9):1327–34.
Fan X, Zhao S, Yu C, Wu D, Yan Z, Fan L, et al. Exome sequencing reveals genetic architecture in patients with isolated or syndromic short stature. J Genet Genomics. 2021;48(5):396–402.
Rappold GA, Fukami M, Niesler B, Schiller S, Z-umkeller W, Bettendorf M, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002;87(3):1402–6.
Argente J. Challenges in the management of short stature. Horm Res Paediatr. 2016;85(1):2–10.
Grunauer M, Jorge AAL. Genetic short stature. Growth Horm IGF Res. 2018;38:29–33.
Guo L, Park J, Yi E, Marchi E, Hsieh T, Kibalnyk Y et al. KBG syndrome: videoconferencing and use of artificial intelligence driven facial phenotyping in 25 new patients. Eur J Hum Genet. 2022;1–11.
Ranke MB, Wit JM. Growth hormone-past, present and future. Nat Rev Endocrinol. 2018;14(5):285–300.
Collett-Solberg PF, Jorge AAL, Boguszewski MCS, Miller BS, Choong CSY, Cohen P, et al. Growth hormone therapy in children; research and practice-a review. Growth Horm Igf Res. 2019;44:20–32.
Dauber A. Genetic testing for the child with short stature-has the time come to change our diagnostic paradigm? J Clin Endocr Metab. 2019;104(7):2766–9.
Muthuvel G, Dauber A, Alexandrou E, Tyzinski L, Andrew M, Hwa V, et al. Treatment of short stature in aggrecan-deficient patients with recombinant human growth hormone: 1-year response. J Clin Endocr Metab. 2022;107(5):E2103–9.
van der Steen M, Pfundt R, Maas SJWH, Waarde WMBV, Odink RJ, Hokken-Koelega ACS. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J Clin Endocr Metab. 2017;102(5):1458–67.
Ke XA, Liang HT, Miao H, Yang HB, Wang LJ, Gong FY, et al. Clinical characteristics of short-stature patients with an NPR2 mutation and the therapeutic response to rhGH. J Clin Endocr Metab. 2021;106(2):431–41.
Plachy L, Dusatkova P, Maratova K, Petruzelkova L, Zemkova D, Elblova L, et al. NPR2 variants are frequent among children with familiar short stature and respond well to growth hormone therapy. J Clin Endocr Metab. 2020;105(3):E746–52.
Vasques GA, Funari MFA, Ferreira FM, Aza-Carmona M, Sentchordi-Montane L, Barraza-Garcia J, et al. IHH gene mutations causing short stature with nonspecific skeletal abnormalities and response to growth hormone therapy. J Clin Endocr Metab. 2018;103(2):604–14.
Noll JE, Jeffery J, Al-Ejeh F, Kumar R, Khanna KK, Callen DF, et al. Mutant p53 drives multinucleation and invasion through a process that is suppressed by ANKRD11. Oncogene. 2012;31(23):2836–48.
Child CJ, Zimmermann AG, Chrousos GP, Cummings E, Deal CL, Hasegawa T, et al. Safety outcomes during pediatric GH therapy: final results from the prospective GeNeSIS observational program. J Clin Endocr Metab. 2019;104(2):379–89.
Child CJ, Zimmermann AG, Jia N, Robison LL, Bramswig JH, Blum WF. Assessment of primary cancer incidence in growth hormone-treated children: comparison of a multinational prospective observational study with population databases. Horm Res Paediat. 2016;85(3):198–206.
Wilton P, Mattsson AF, Darendeliler F. Growth hormone treatment in children is not associated with an increase in the incidence of cancer: experience from KIGS (Pfizer International Growth Database). J Pediatr. 2010;157(2):265–70.
Behnert A, Auber B, Steinemann D, Frühwald MC, Huisinga C, Hussein K, et al. KBG syndrome patient due to 16q24. 3 microdeletion presenting with a paratesticular rhabdoid tumor: coincidence or cancer predisposition? Am J Med Genet A. 2018;176(6):1449–54.
Isrie M, Hendriks Y, Gielissen N, Sistermans EA, Willemsen MH, Peeters H, et al. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur J Hum Genet. 2012;20(2):131–3.
Lui JC. Home for a rest: stem cell niche of the postnatal growth plate. J Endocrinol. 2020;246(1):R1–11.
Hallett SA, Matsushita Y, Ono W, Sakagami N, Mizuhashi K, Tokavanich N, et al. Chondrocytes in the resting zone of the growth plate are maintained in a wnt-inhibitory environment. Elife. 2021;10:e64513.
Morris SA. Single-cell RNA-seq steps up to the growth plate. Trends Biotechnol. 2016;34(7):525–27.
Hallett SA, Ono W, Ono N. Growth plate chondrocytes: skeletal development, growth and beyond. Int J Mol Sci. 2019;20(23):6009.
Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. MECHANISMS IN ENDOCRINOLOGY: novel genetic causes of short stature. Eur J Endocrinol. 2016;174(4):R145–73.
Baron J, Savendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015;11(12):735–46.
Faienza MF, Chiarito M, Brunetti G, D’Amato G. Growth plate gene involment and isolated short stature. Endocrine. 2021;71(1):28–34.
Borjesson AE, Lagerquist MK, Windahl SH, Ohlsson C. The role of estrogen receptor alpha in the regulation of bone and growth plate cartilage. Cell Mol Life Sci. 2013;70(21):4023–37.
Negishi Y, Ui N, Nakajima M, Kawashima K, Maruyama K, Takizawa T, et al. p21Cip-1/SDI-1/WAF-1 gene is involved in chondrogenic differentiation of ATDC5 cells in vitro. J Biol Chem. 2001;276(35):33249–56.
Yao Y, Wang Y. ATDC5: an excellent in vitro model cell line for skeletal development. J Cell Biochem. 2013;114(6):1223–9.
Cheng X, Li P, Wang G, Yan Y, Li K. Microbiota-derived lipopolysaccharide retards chondrocyte hypertrophy in the growth plate through elevating Sox9 expression. J Cell Physiol. 2019;234(3):2593–605.
Dong X, Xu X, Yang C, Luo Y, Wu Y, Wang J. USP7 regulates the proliferation and differentiation of ATDC5 cells through the Sox9-PTHrP-PTH1R axis. Bone. 2021;143:115714.
Download references
Acknowledgements
Not applicable.
This work was supported by Research Fund for Academician Lin He New Medicine (JYHL2019FZD01) and the PhD Research Foundation of Affiliated Hospital of Jining Medical University (2018-BS-007), and was partly supported by Shandong Traditional Chinese Medicine Science and Technology Development Plans Project (2019 − 0486).
Author information
Authors and affiliations.
Department of Endocrinology, Genetics and Metabolism, Affiliated Hospital of Jining Medical University, Jining, Shandong, 272029, China
Dongye He, Mei Zhang, Yanying Li, Fupeng Liu & Bo Ban
Medical Research Center, Affiliated Hospital of Jining Medical University, Jining, China
Dongye He, Fupeng Liu & Bo Ban
Chinese Research Center for Behavior Medicine in Growth and Development, Jining, China
Mei Zhang, Yanying Li & Bo Ban
You can also search for this author in PubMed Google Scholar
Contributions
DYH performed the literature search and wrote the manuscript. MZ, YYL and FPL performed the literature search and collected ANKRD11 variants from ClinVar database. BB provided guidance on the data collection and critically revised the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding authors
Correspondence to Dongye He or Bo Ban .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
The autors declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary material 2, supplementary material 3, rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
He, D., Zhang, M., Li, Y. et al. Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search. Orphanet J Rare Dis 19 , 292 (2024). https://doi.org/10.1186/s13023-024-03301-y
Download citation
Received : 03 April 2023
Accepted : 05 August 2024
Published : 12 August 2024
DOI : https://doi.org/10.1186/s13023-024-03301-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- ANKRD11 gene
- KBG syndrome
- Hotspot variants
- Short stature
- Growth hormone treatment
- Growth plate development
Orphanet Journal of Rare Diseases
ISSN: 1750-1172
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
Information
- Author Services
Initiatives
You are accessing a machine-readable page. In order to be human-readable, please install an RSS reader.
All articles published by MDPI are made immediately available worldwide under an open access license. No special permission is required to reuse all or part of the article published by MDPI, including figures and tables. For articles published under an open access Creative Common CC BY license, any part of the article may be reused without permission provided that the original article is clearly cited. For more information, please refer to https://www.mdpi.com/openaccess .
Feature papers represent the most advanced research with significant potential for high impact in the field. A Feature Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for future research directions and describes possible research applications.
Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive positive feedback from the reviewers.
Editor’s Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world. Editors select a small number of articles recently published in the journal that they believe will be particularly interesting to readers, or important in the respective research area. The aim is to provide a snapshot of some of the most exciting work published in the various research areas of the journal.
Original Submission Date Received: .
- Active Journals
- Find a Journal
- Proceedings Series
- For Authors
- For Reviewers
- For Editors
- For Librarians
- For Publishers
- For Societies
- For Conference Organizers
- Open Access Policy
- Institutional Open Access Program
- Special Issues Guidelines
- Editorial Process
- Research and Publication Ethics
- Article Processing Charges
- Testimonials
- Preprints.org
- SciProfiles
- Encyclopedia
Article Menu

- Subscribe SciFeed
- Recommended Articles
- Google Scholar
- on Google Scholar
- Table of Contents
Find support for a specific problem in the support section of our website.
Please let us know what you think of our products and services.
Visit our dedicated information section to learn more about MDPI.
JSmol Viewer
A bi-objective model for the multi-period inventory-based reverse logistics network: a case study from an automobile component distribution network.

1. Introduction
- To optimize the transportation system in the ISACO company.
- To cut down transportation costs.
- To increase customer satisfaction by increasing the supply of customer demands.
- To allow the customers to return unused parts (which are not used by customers due to seasonal variations or environmental changes and market fluctuations.
- To collect and dispose or recycle the stock parts.
2. Literature Review
2.1. a review of the literature on distribution systems in supply chain management, 2.2. a review of the literature on green logistics in supply chain management, 3. materials and methods.
- Very high transportation costs induced by long round-trip distances.
- High costs imposed on the company as a result of vehicle breakdown.
- Frequent troubles related to timely goods delivery (e.g., the cities located far from Tehran, the chances are high that the goods do not reach on time).
- To benefit from the full capacity of cars, it is required that the amount of the ordered goods reach a certain quantity and then the goods be delivered to the representatives, which leads to dissatisfaction among the representatives and losing the competitive market.
- The lack of order and prioritization in the current system.
- Not considering different scenarios in decision making.
- Not being able to return unused or low-use parts by the representatives.
- The lack of an integrated system for receiving scrap parts.
- Not able to implement strategic planning.
- Some of the expected merits of the new system are the following:
- Reducing the costs resulting from redundant transportation.
- Increasing the representatives’ satisfaction level due to goods’ timely delivery and increasing the power to supply the demanded goods and the possibility of returning low-use parts to the representative.
- Systematizing transportation system which curbs other nuisances.
- Increasing the flexibility of the system.
- Decreasing the risks such as the sensitive parts becoming faulty during long transportation or the possibility of vehicle breakdowns that impose losses on the company.
- Building regional warehouses and reducing the heavy costs of the central warehouse.
- Controlling the system better and the potential to constantly improve.
5. Discussion and Conclusions
- Employing a multi-period model along with the power of inventory management so that it leads to reduced costs and increased revenue.
- With respect to the variety of available products, the number of product groups should be increased and included in the proposed model.
- Reducing the time of ordering periods to better use the multi-period model, supplying faster and more up-to-date customer demands in the year, and removing the barriers of the inventory cost increase through modeling and making decisions at the tactical and operational level.
- Raising the number of customers and applying the proposed model to the actual number of customers. It is worth mentioning that in this model, they were integrated into the provincial centers to facilitate the modeling of customer demand.
- Constructing regional warehouses in the locations suggested by the model outputs considering the construction cost and setting up and storing the goods in these warehouses.
- Launching the central warehouse number 2 when its effectiveness gets approved in all the models to properly benefit from it.
- Regularly controlling the proposed performance evaluation indices considering the possibility of changing the supply or demand pattern and making suitable decisions accordingly.
- Investigating the demand pattern in various time periods and the possibility of presenting a supplementary model for the probability mode of demand.
- Investigating the profit from waste recycling.
- Investigating the benefits of the brand’s mental image in terms of compliance with environmental issues.
- Considering production issues in the supply chain and distribution system.
- Including the demand of the different classes of customers in the distribution system and locating facilities; accordingly, in other words, assessing the effect of marketing decisions on the strategic macro-decisions of facility location.
- Considering other location benchmarks.
- Determining the order supply deadline for all sorts of goods orders and programming to supply them within the deadline and its effect on facility location problems.
- Considering other objective functions like social aspects, employment rates, and environmental impacts according to the priorities of managers and decision-makers.
Author Contributions
Data availability statement, conflicts of interest.
- Kamalahmadi, M.; Parast, M.M. A review of the literature on the principles of enterprise and supply chain resilience: Major findings and directions for future research. Int. J. Prod. Econ. 2016 , 171 , 116–133. [ Google Scholar ] [ CrossRef ]
- Wang, G.; Gunasekaran, A.; Ngai, E.W.T.; Papadopoulos, T. Big data analytics in logistics and supply chain management: Certain investigations for research and applications. Int. J. Prod. Econ. 2016 , 176 , 98–110. [ Google Scholar ] [ CrossRef ]
- Alshubiri, F. The Impact of Green Logistics-Based Activities on the Sustainable Monetary Expansion Indicators of Oman. J. Ind. Eng. Manag. 2017 , 10 , 389–405. [ Google Scholar ] [ CrossRef ]
- Sheikh Azadi, A.H.; Shamsi Nesary, V.; Kebriyaii, O.; Khalilzadeh, M.; Antucheviciene, J. Design of a Green Supply Chain Based on the Kano Model Considering Pricing. Sustainability 2023 , 15 , 13038. [ Google Scholar ] [ CrossRef ]
- Liou, J.J.H.; Tamošaitienė, J.; Zavadskas, E.K.; Tzeng, G.-H. New hybrid COPRAS-G MADM Model for improving and selecting suppliers in green supply chain management. Int. J. Prod. Res. 2015 , 54 , 114–134. [ Google Scholar ] [ CrossRef ]
- Scott, J.; Ho, W.; Dey, P.K.; Talluri, S. A decision support system for supplier selection and order allocation in stochastic, multi-stakeholder and multi-criteria environments. Int. J. Prod. Econ. 2015 , 166 , 226–237. [ Google Scholar ] [ CrossRef ]
- Dweiri, F.; Kumar, S.; Khan, S.A.; Jain, V. Designing an integrated AHP based decision support system for supplier selection in automotive industry. Expert Syst. Appl. 2016 , 62 , 273–283. [ Google Scholar ] [ CrossRef ]
- Mogeni, L.M.; Kiarie, D.M. Effect of Green Logistics Practices on Performance of Supply Chains in Multinational Organizations in Kenya. Ind. Eng. Lett. 2016 , 6 , 40–50. [ Google Scholar ]
- Muangpan, T.; Chaowarat, M.; Neamvonk, J. The key activities of green logistics management in the Thai automotive industry. J. Arts Sci. Commer. 2016 , 7 , 1–11. [ Google Scholar ] [ CrossRef ]
- Brusset, X.; Teller, C. Supply chain capabilities, risks, and resilience. Int. J. Prod. Econ. 2017 , 184 , 59–68. [ Google Scholar ] [ CrossRef ]
- Ribeiro, J.P.; Barbosa-Povoa, A. Supply Chain Resilience: Definitions and quantitative modelling approaches. Comput. Ind. Eng. 2018 , 115 , 109–122. [ Google Scholar ] [ CrossRef ]
- Heidari, A.; Imani, D.M.; Khalilzadeh, M. A hub location model in the sustainable supply chain considering customer segmentation. J. Eng. Des. Technol. 2021 , 19 , 1387–1420. [ Google Scholar ] [ CrossRef ]
- Eydi, A.; Fazayeli, S.; Ghafouri, H. Multi-period configuration of forward and reverse integrated supply chain networks through transport mode. Sci. Iran. 2020 , 27 , 935–955. [ Google Scholar ] [ CrossRef ]
- Antucheviciene, J.; Jafarnejad, A.; Amoozad Mahdiraji, H.; Razavi Hajiagha, S.H.; Kargar, A. Robust Multi-Objective Sustainable Reverse Supply Chain Planning: An Application in the Steel Industry. Symmetry 2020 , 12 , 594. [ Google Scholar ] [ CrossRef ]
- Gholamian, N.; Mahdavi, I.; Mahdavi-Amiri, N.; Tavakkoli-Moghaddam, R. Hybridization of an interactive fuzzy methodology with a lexicographic min-max approach for optimizing a multi-period multi-product multi-echelon sustainable closed-loop supply chain network. Comput. Ind. Eng. 2021 , 158 , 107282. [ Google Scholar ] [ CrossRef ]
- Alinezhad, M.; Mahdavi, I.; Hematian, M. A fuzzy multi-objective optimization model for sustainable closed-loop supply chain network design in food industries. Environ. Dev. Sustain. 2022 , 24 , 8779–8806. [ Google Scholar ] [ CrossRef ]
- Pazhani, S.; Mendoza, A.; Nambirajan, R.; Narendran, T.T.; Ganesh, K.; Olivares-Benitez, E. Multi-period multi-product closed loop supply chain network design: A relaxation approach. Comput. Ind. Eng. 2021 , 155 , 107191. [ Google Scholar ] [ CrossRef ]
- Nazari-Ghanbarloo, V.; Ghodratnama, A. Optimizing a robust tri-objective multi-period reliable supply chain network considering queuing system and operational and disruption risks. Oper. Res. 2021 , 21 , 1963–2020. [ Google Scholar ] [ CrossRef ]
- Rabe, M.; Gonzalez-Feliu, J.; Chicaiza-Vaca, J.; Tordecilla, R.D. Simulation-Optimization Approach for Multi-Period Facility Location Problems with Forecasted and Random Demands in a Last-Mile Logistics Application. Algorithms 2021 , 14 , 41. [ Google Scholar ] [ CrossRef ]
- Zarrat Dakhely Parast, Z.; Haleh, H.; Avakh Darestani, S. Green reverse supply chain network design considering location-routing-inventory decisions with simultaneous pickup and delivery. Environ. Sci. Pollut. Res. 2021 . [ Google Scholar ] [ CrossRef ]
- Abdolazimi, O.; Bahrami, F.; Shishebori, D. A multi-objective closed-loop supply chain network design problem under parameter uncertainty: Comparison of exact methods. Environ. Dev. Sustain. 2022 , 24 , 10768–10802. [ Google Scholar ] [ CrossRef ]
- Maliki, F.; Souier, M.; Dahane, M. A multi-objective optimization model for a multi-period mobile facility location problem with environmental and disruption considerations. Ann. Oper. Res. 2022 , 1–26. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Gupta, S.; Vijaygargy, L.; Sarkar, B. A bi-objective integrated transportation and inventory management under a supply chain network considering multiple distribution networks. RAIRO Oper. Res. 2022 , 56 , 3991–4022. [ Google Scholar ] [ CrossRef ]
- Bortolini, M.; Calabrese, F.; Gabriele Galizia, F.; Mora, C. A three-objective optimization model for mid-term sustainable supply chain network design. Comput. Ind. Eng. 2022 , 168 , 108131. [ Google Scholar ] [ CrossRef ]
- Mogale, D.G.; De, A.; Ghadge, A.; Aktas, E. Multi-objective modelling of sustainable closed-loop supply chain network with price-sensitive demand and consumer’s incentives. Comput. Ind. Eng. 2022 , 168 , 108105. [ Google Scholar ] [ CrossRef ]
- Al-Refaie, A.; Kokash, T. Optimization of sustainable reverse logistics network with multi-objectives under uncertainty. J. Remanuf. 2023 , 13 , 1–23. [ Google Scholar ] [ CrossRef ]
- Meidute-Kavaliauskiene, I.; Yıldırım, F.; Ghorbani, S.; Činčikaitė, R. The Design of a Multi-Period and Multi-Echelon Perishable Goods Supply Network under Uncertainty. Sustainability 2022 , 14 , 2472. [ Google Scholar ] [ CrossRef ]
- Allehashemi, T.; Hassanzadeh Amin, S.; Zolfaghari, S. A proposed multi-objective model for cellphone closed-loop supply chain optimization based on fuzzy QFD. Expert Syst. Appl. 2022 , 210 , 118577. [ Google Scholar ] [ CrossRef ]
- Abdi, F.; Farughi, H.; Sadeghi, H.; Arkat, J. Location-inventory-reliability optimisation problem in a multi-objective multi-period three-level supply chain network with stochastic demand. Eur. J. Ind. Eng. 2023 , 17 , 479–528. [ Google Scholar ] [ CrossRef ]
- Arab Momeni, M.; Jain, V.; Bagheri, M. A Multi-Objective Model for Designing a Sustainable Closed-Loop Supply Chain Logistics Network. Logistics 2024 , 8 , 29. [ Google Scholar ] [ CrossRef ]
- Varshney, N.; Gupta, S.; Ahmed, A. Addressing uncertainty in closed-loop supply chain networks: A multi-objective approach to integrated production and transportation problems. J. Model. Manag. 2024 , in press . [ Google Scholar ] [ CrossRef ]
- Xia, H.; Chen, Z.; Milisavljevic-Syed, J.; Salonitis, K. Uncertain programming model for designing multi-objective reverse logistics networks. Clean. Logist. Supply Chain 2024 , 11 , 100155. [ Google Scholar ] [ CrossRef ]
Click here to enlarge figure
| α | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | |||
| β | 0 | Z1 | 4.82 × 10 | 3.50 × 10 | 2.19 × 10 | 1.90 × 10 | 1.34 × 10 | 9.49 × 10 | 5.34 × 10 | 0.00 × 10 | 0.00 × 10 |
| Z2 | 1 | 0.95 | 0.8368985 | 0.8 | 0.6857143 | 0.5714286 | 0.3811921 | 0 | 0 | ||
| 0.1 | Z1 | 4.82 × 10 | 3.64 × 10 | 3.19 × 10 | 2.27 × 10 | 1.98 × 10 | 1.64 × 10 | 1.18 × 10 | 1.10 × 10 | 9.38 × 10 | |
| Z2 | 1 | 0.955 | 0.9228571 | 0.8264286 | 0.775 | 0.6464286 | 0.5371429 | 0.4857143 | 0.2649475 | ||
| 0.2 | Z1 | 4.82 × 10 | 3.78 × 10 | 3.37 × 10 | 2.59 × 10 | 2.33 × 10 | 1.87 × 10 | 1.62 × 10 | 1.55 × 10 | 1.43 × 10 | |
| Z2 | 1 | 0.96 | 0.9314286 | 0.8457143 | 0.8 | 0.6857143 | 0.5885714 | 0.5428571 | 0.3942857 | ||
| 0.3 | Z1 | 4.82 × 10 | 4.11 × 10 | 3.55 × 10 | 2.90 × 10 | 2.68 × 10 | 2.28 × 10 | 2.06 × 10 | 2.00 × 10 | 1.90 × 10 | |
| Z2 | 1 | 0.975 | 0.94 | 0.865 | 0.825 | 0.725 | 0.64 | 0.6 | 0.47 | ||
| 0.4 | Z1 | 4.82 × 10 | 4.21 × 10 | 3.73 × 10 | 3.31 × 10 | 3.07 × 10 | 2.69 × 10 | 2.50 × 10 | 2.44 × 10 | 2.36 × 10 | |
| Z2 | 1 | 0.9785714 | 0.9485714 | 0.9014286 | 0.8585714 | 0.7642857 | 0.6914286 | 0.6571429 | 0.5457143 | ||
| 0.5 | Z1 | 4.82 × 10 | 4.31 × 10 | 3.92 × 10 | 3.56 × 10 | 3.36 × 10 | 3.09 × 10 | 2.94 × 10 | 2.88 × 10 | 2.81 × 10 | |
| Z2 | 1 | 0.9821429 | 0.9571429 | 0.9178571 | 0.8821429 | 0.8142857 | 0.7607143 | 0.7214286 | 0.6214286 | ||
| 0.6 | Z1 | 4.82 × 10 | 4.41 × 10 | 4.10 × 10 | 3.82 × 10 | 3.65 × 10 | 3.43 × 10 | 3.32 × 10 | 3.27 × 10 | 3.21 × 10 | |
| Z2 | 1 | 0.9857143 | 0.9657143 | 0.9342857 | 0.9057143 | 0.8514286 | 0.8085714 | 0.7771429 | 0.6971429 | ||
| 0.7 | Z1 | 4.82 × 10 | 4.51 × 10 | 4.28 × 10 | 4.07 × 10 | 3.94 × 10 | 3.78 × 10 | 3.69 × 10 | 3.66 × 10 | 3.61 × 10 | |
| Z2 | 1 | 0.9892857 | 0.9742857 | 0.9507143 | 0.9292857 | 0.8885714 | 0.8564286 | 0.8328571 | 0.7728571 | ||
| 0.8 | Z1 | 4.82 × 10 | 4.62 × 10 | 4.46 × 10 | 4.32 × 10 | 4.24 × 10 | 4.13 × 10 | 4.07 × 10 | 4.05 × 10 | 4.01 × 10 | |
| Z2 | 1 | 0.9928571 | 0.9828571 | 0.9671429 | 0.9528571 | 0.9257143 | 0.9042857 | 0.8885714 | 0.8485714 | ||
| 0.9 | Z1 | 4.82 × 10 | 4.72 × 10 | 4.64 × 10 | 4.57 × 10 | 4.53 × 10 | 4.47 × 10 | 4.44 × 10 | 4.43 × 10 | 4.42 × 10 | |
| Z2 | 1 | 0.9964286 | 0.9914286 | 0.9832714 | 0.9764286 | 0.9628571 | 0.9521429 | 0.9442857 | 0.9242857 | ||
| 1 | Z1 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | 4.82 × 10 | |
| Z2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Variable Title | Value |
|---|---|
| Z | 3.32 × 10 |
| Z | 80% |
| V(1,2) | (1,1) |
| U(1,2,3,4,5,6,7,8) | (0,0,0,0,0,1,0,1) |
| α | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | |||
| β | 0 | Z1 | 4.84 × 10 | 4.70 × 10 | 4.52 × 10 | 4.20 × 10 | 3.68 × 10 | 3.54 × 10 | 3.41 × 10 | 3.15 × 10 | 2.65 × 10 |
| Z2 | 1 | 0.9928571 | 0.9785714 | 0.9428571 | 0.8494217 | 0.8142857 | 0.7571429 | 0.5928571 | 0 | ||
| 0.1 | Z1 | 4.84 × 10 | 4.72 × 10 | 4.55 × 10 | 4.27 × 10 | 3.82 × 10 | 3.70 × 10 | 3.58 × 10 | 3.35 × 10 | 3.31 × 10 | |
| Z2 | 1 | 0.9935714 | 0.9807143 | 0.9485714 | 0.8624167 | 0.8328571 | 0.7814286 | 0.6335714 | 0.5821429 | ||
| 0.2 | Z1 | 4.84 × 10 | 4.73 × 10 | 4.58 × 10 | 4.22 × 10 | 4.15 × 10 | 3.87 × 10 | 3.76 × 10 | 3.55 × 10 | 0.5821429 | |
| Z2 | 1 | 0.9942857 | 0.9828571 | 0.9371429 | 0.9257143 | 0.8514286 | 0.8057143 | 0.6742857 | 0.6285714 | ||
| 0.3 | Z1 | 4.84 × 10 | 4.74 × 10 | 4.62 × 10 | 4.30 × 10 | 4.24 × 10 | 4.14 × 10 | 3.94 × 10 | 3.75 × 10 | 3.72 × 10 | |
| Z2 | 1 | 0.995 | 0.985 | 0.945 | 0.935 | 0.91 | 0.83 | 0.715 | 0.675 | ||
| 0.4 | Z1 | 4.84 × 10 | 4.76 × 10 | 4.65 × 10 | 4.37 × 10 | 4.33 × 10 | 4.24 × 10 | 4.14 × 10 | 3.98 × 10 | 3.93 × 10 | |
| Z2 | 1 | 0.9957143 | 0.9871429 | 0.9528571 | 0.9442857 | 0.9228571 | 0.88 | 0.7814286 | 0.7214286 | ||
| 0.5 | Z1 | 4.84 × 10 | 4.77 × 10 | 4.68 × 10 | 4.45 × 10 | 4.41 × 10 | 4.34 × 10 | 4.26 × 10 | 4.12 × 10 | 4.11 × 10 | |
| Z2 | 1 | 0.9964286 | 0.9892857 | 0.9607143 | 0.9535714 | 0.9357143 | 0.9 | 0.8178571 | 0.7928571 | ||
| 0.6 | Z1 | 4.84 × 10 | 4.79 × 10 | 4.71 × 10 | 4.53 × 10 | 4.50 × 10 | 4.44 × 10 | 4.37 × 10 | 4.27 × 10 | 4.26 × 10 | |
| Z2 | 1 | 0.9971429 | 0.9914286 | 0.9685714 | 0.9628571 | 0.9485714 | 0.92 | 0.8542857 | 0.8342857 | ||
| 0.7 | Z1 | 4.84 × 10 | 4.80 × 10 | 4.75 × 10 | 4.61 × 10 | 4.58 × 10 | 4.54 × 10 | 4.49 × 10 | 4.41 × 10 | 4.40 × 10 | |
| Z2 | 1 | 0.9978571 | 0.9935714 | 0.9764286 | 0.9721429 | 0.9614286 | 0.94 | 0.8907143 | 0.8757143 | ||
| 0.8 | Z1 | 4.84 × 10 | 4.82 × 10 | 4.78 × 10 | 4.69 × 10 | 4.67 × 10 | 4.64 × 10 | 4.61 × 10 | 4.56 × 10 | 4.55 × 10 | |
| Z2 | 1 | 0.9985714 | 0.9957143 | 0.9842857 | 0.981286 | 0.9742857 | 0.96 | 0.9271429 | 0.9171429 | ||
| 0.9 | Z1 | 4.84 × 10 | 4.83 × 10 | 4.81 × 10 | 4.77 × 10 | 4.76 × 10 | 4.74 × 10 | 4.73 × 10 | 4.70 × 10 | 4.70 × 10 | |
| Z2 | 1 | 0.9992857 | 0.9978571 | 0.9921429 | 0.9907143 | 0.9871429 | 0.98 | 0.9635714 | 0.9585714 | ||
| 1 | Z1 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | 4.84 × 10 | |
| Z2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Variable Title | Value |
|---|---|
| Z | 3.37 × 10 |
| Z | 92% |
| V(1,2) | (1,1) |
| U(1,2,3,4,5,6,7,8) | (0,0,0,0,0,1,0,1) |
| α | ||||||||
|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.51 × 10 | 1.48 × 10 | 1.37 × 10 | 1.34 × 10 | 1.21 × 10 |
| 1 | 1 | 1 | 0.9228571 | 0.9714286 | 0.9642857 | 0.9150749 | 0.888311 | 0.7168279 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.52 × 10 | 1.50 × 10 | 1.40 × 10 | 1.37 × 10 | 1.26 × 10 |
| 1 | 1 | 1 | 0.9935714 | 0.9742857 | 0.9678571 | 0.9232892 | 0.8982267 | 0.7361599 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.54 × 10 | 1.52 × 10 | 1.43 × 10 | 1.40 × 10 | 1.33 × 10 |
| 1 | 1 | 1 | 0.9942857 | 0.9771429 | 09744286 | 0.9315568 | 0.9085202 | 0.8056308 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 0.62 × 10 | 1.55 × 10 | 1.53 × 10 | 1.46 × 10 | 1.43 × 10 | 1.37 × 10 |
| 1 | 1 | 1 | 0.995 | 0.98 | 0.975 | 0.939905 | 0.919635 | 0.8278087 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 0.62 × 10 | 1.57 × 10 | 1.55 × 10 | 1.49 × 10 | 1.47 × 10 | 1.41 × 10 |
| 1 | 1 | 1 | 0.9957143 | 0.9828571 | 0.9785714 | 0.9482679 | 0.9307498 | 0.8485393 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.63 × 10 | 1.58 × 10 | 1.57 × 10 | 1.52 × 10 | 1.50 × 10 | 1.45 × 10 |
| 1 | 1 | 1 | 0.9664286 | 0.9857143 | 0.9821429 | 0.9566604 | 0.9418646 | 0.8732143 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.63 × 10 | 1.57 × 10 | 1.48 × 10 | 1.43 × 10 | 1.41 × 10 | 1.39 × 10 |
| 1 | 1 | 1 | 0.9971429 | 0.9885714 | 0.9857143 | 0.9654133 | 0.9530132 | 0.8985714 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.64 × 10 | 1.61 × 10 | 1.60 × 10 | 1.57 × 10 | 1.56 × 10 | 1.53 × 10 |
| 1 | 1 | 1 | 0.9978571 | 0.9914286 | 0.9892857 | 0.9742857 | 0.964634 | 0.9238566 |
| 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.64 × 10 | 1.62 × 10 | 1.62 × 10 | 1.61 × 10 | 1.59 × 10 | 1.57 × 10 |
| 1 | 1 | 1 | 0.9985714 | 0.9942857 | 0.9928571 | 0.9885714 | 0.97625 | 0.9489286 |
| 1.65 × 10 | 0.2 | 0.3 | 0.4 | 1.64 × 10 | 1.64 × 10 | 1.63 × 10 | 1.63 × 10 | 1.62 × 10 |
| 1 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 0.9971429 | 0.9964286 | 0.9942857 | 0.9895536 | 0.9785714 |
| 1.65 × 10 | 1 | 1 | 0.9228571 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 |
| 1 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1 | 1 | 1 | 1 | 1 |
| Variable Title | Value |
|---|---|
| Z | 1.42 × 10 |
| Z | 94% |
| V(1,2) | (1,1) |
| U(1,2,3,4,5,6,7,8) | (1,0,1,1,0,1,0,1) |
| α | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 | 0.7 | 0.8 | 0.9 | |||
| β | 0 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.51 × 10 | 1.48 × 10 | 1.37 × 10 | 1.34 × 10 | 1.21 × 10 |
| Z2 | 1 | 1 | 1 | 0.9228571 | 0.9714286 | 0.9642857 | 0.9150749 | 0.888311 | 0.7168279 | ||
| 0.1 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.52 × 10 | 1.50 × 10 | 1.40 × 10 | 1.37 × 10 | 1.26 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9935714 | 0.9742857 | 0.9678571 | 0.9232892 | 0.8982267 | 0.7361599 | ||
| 0.2 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.61 × 10 | 1.54 × 10 | 1.52 × 10 | 1.43 × 10 | 1.40 × 10 | 1.33 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9942857 | 0.9771429 | 0.9744286 | 0.9315568 | 0.9085202 | 0.8056308 | ||
| 0.3 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 0.62 × 10 | 1.55 × 10 | 1.53 × 10 | 1.46 × 10 | 1.43 × 10 | 1.37 × 10 | |
| Z2 | 1 | 1 | 1 | 0.995 | 0.98 | 0.975 | 0.939905 | 0.919635 | 0.8278087 | ||
| 0.4 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 0.62 × 10 | 1.57 × 10 | 1.55 × 10 | 1.49 × 10 | 1.47 × 10 | 1.41 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9957143 | 0.9828571 | 0.9785714 | 0.9482679 | 0.9307498 | 0.8485393 | ||
| 0.5 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.63 × 10 | 1.58 × 10 | 1.57 × 10 | 1.52 × 10 | 1.50 × 10 | 1.45 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9664286 | 0.9857143 | 0.9821429 | 0.9566604 | 0.9418646 | 0.8732143 | ||
| 0.6 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.63 × 10 | 1.57 × 10 | 1.48 × 10 | 1.43 × 10 | 1.41 × 10 | 1.39 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9971429 | 0.9885714 | 0.9857143 | 0.9654133 | 0.9530132 | 0.8985714 | ||
| 0.7 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.64 × 10 | 1.61 × 10 | 1.60 × 10 | 1.57 × 10 | 1.56 × 10 | 1.53 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9978571 | 0.9914286 | 0.9892857 | 0.9742857 | 0.964634 | 0.9238566 | ||
| 0.8 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.64 × 10 | 1.62 × 10 | 1.62 × 10 | 1.61 × 10 | 1.59 × 10 | 1.57 × 10 | |
| Z2 | 1 | 1 | 1 | 0.9985714 | 0.9942857 | 0.9928571 | 0.9885714 | 0.97625 | 0.9489286 | ||
| 0.9 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.64 × 10 | 1.64 × 10 | 1.63 × 10 | 1.63 × 10 | 1.62 × 10 | |
| Z2 | 1 | 1 | 1 | 1 | 0.9971429 | 0.9964286 | 0.9942857 | 0.9895536 | 0.9785714 | ||
| 1 | Z1 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | 1.65 × 10 | |
| Z2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Variable Title | Value |
|---|---|
| Z | 1.43 × 10 |
| Z | 96% |
| V(1,2) | (1,1) |
| U(1,2,3,4,5,6,7,8) | (1,0,1,1,0,1,0,1) |
| Criterion Illustration | Criterion Components | Basic Model | Basic Model with Inventory Management | Multi-Period Basic Model with Inventory Management | Multi-Period Basic Model with Inventory Management and Green Logistics |
|---|---|---|---|---|---|
| Overall Satisfaction of Customers | 85% | 92% | 94% | 96% | |
| Total Costs | 3.32 × 10 | 4.37 × 10 | 1.42 × 10 | 1.43 × 10 |
| The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Share and Cite
Khalilzadeh, M.; Antucheviciene, J.; Božanić, D. A Bi-Objective Model for the Multi-Period Inventory-Based Reverse Logistics Network: A Case Study from an Automobile Component Distribution Network. Systems 2024 , 12 , 299. https://doi.org/10.3390/systems12080299
Khalilzadeh M, Antucheviciene J, Božanić D. A Bi-Objective Model for the Multi-Period Inventory-Based Reverse Logistics Network: A Case Study from an Automobile Component Distribution Network. Systems . 2024; 12(8):299. https://doi.org/10.3390/systems12080299
Khalilzadeh, Mohammad, Jurgita Antucheviciene, and Darko Božanić. 2024. "A Bi-Objective Model for the Multi-Period Inventory-Based Reverse Logistics Network: A Case Study from an Automobile Component Distribution Network" Systems 12, no. 8: 299. https://doi.org/10.3390/systems12080299
Article Metrics
Article access statistics, further information, mdpi initiatives, follow mdpi.
Subscribe to receive issue release notifications and newsletters from MDPI journals
IMAGES
COMMENTS
This is why the literature review as a research method is more relevant than ever. Traditional literature reviews often lack thoroughness and rigor and are conducted ad hoc, rather than following a specific methodology. Therefore, questions can be raised about the quality and trustworthiness of these types of reviews.
9.3. Types of Review Articles and Brief Illustrations. EHealth researchers have at their disposal a number of approaches and methods for making sense out of existing literature, all with the purpose of casting current research findings into historical contexts or explaining contradictions that might exist among a set of primary research studies conducted on a particular topic.
A literature review can be a part of a research paper or scholarly article, usually falling after the introduction and before the research methods sections. In these cases, the lit review just needs to cover scholarship that is important to the issue you are writing about; sometimes it will also cover key sources that informed your research ...
Examples of literature reviews. Step 1 - Search for relevant literature. Step 2 - Evaluate and select sources. Step 3 - Identify themes, debates, and gaps. Step 4 - Outline your literature review's structure. Step 5 - Write your literature review.
A literature review is defined as "a critical analysis of a segment of a published body of knowledge through summary, classification, and comparison of prior research studies, reviews of literature, and theoretical articles." (The Writing Center University of Winconsin-Madison 2022) A literature review is an integrated analysis, not just a summary of scholarly work on a specific topic.
The literature review represents a method because the literature reviewer chooses from an array of strategies and procedures for identifying, recording, understanding, meaning-making, and transmitting information pertinent to a topic of interest. Moreover,
This book includes steps for students and experienced scholars, with discussion of a variety of literature review types. Conducting research literature reviews:From the Internet to Paper (Fink, 2019). Available resources include Chapters 1 and 2. This edition includes recommendations for organizing literature reviews using online resources.
Literature Review is a comprehensive survey of the works published in a particular field of study or line of research, usually over a specific period of time, in the form of an in-depth, critical bibliographic essay or annotated list in which attention is drawn to the most significant works. Also, we can define a literature review as the ...
Literature reviews establish the foundation of academic inquires. However, in the planning field, we lack rigorous systematic reviews. In this article, through a systematic search on the methodology of literature review, we categorize a typology of literature reviews, discuss steps in conducting a systematic literature review, and provide suggestions on how to enhance rigor in literature ...
A literature review is a review and synthesis of existing research on a topic or research question. A literature review is meant to analyze the scholarly literature, make connections across writings and identify strengths, weaknesses, trends, and missing conversations. ... SAGE Research Methods is the ultimate methods library with more than ...
A literature review involves researching, reading, analyzing, evaluating, and summarizing scholarly literature (typically journals and articles) about a specific topic. The results of a literature review may be an entire report or article OR may be part of a article, thesis, dissertation, or grant proposal.
Literature Review. A literature review is a discussion of the literature (aka. the "research" or "scholarship") surrounding a certain topic. A good literature review doesn't simply summarize the existing material, but provides thoughtful synthesis and analysis. The purpose of a literature review is to orient your own work within an existing ...
Literature Review and Research Design by Dave Harris This book looks at literature review in the process of research design, and how to develop a research practice that will build skills in reading and writing about research literature--skills that remain valuable in both academic and professional careers. Literature review is approached as a process of engaging with the discourse of scholarly ...
A literature review is an integrated analysis-- not just a summary-- of scholarly writings and other relevant evidence related directly to your research question.That is, it represents a synthesis of the evidence that provides background information on your topic and shows a association between the evidence and your research question.
A Rapid Literature Review (RLR) is the fastest type of literature review which makes use of a streamlined approach for synthesizing literature summaries, offering a quicker and more focused alternative to traditional systematic reviews. Despite employing identical research methods, it often simplifies or omits specific steps to expedite the ...
Method details Overview. A Systematic Literature Review (SLR) is a research methodology to collect, identify, and critically analyze the available research studies (e.g., articles, conference proceedings, books, dissertations) through a systematic procedure [12].An SLR updates the reader with current literature about a subject [6].The goal is to review critical points of current knowledge on a ...
Types of Literature Review are as follows: Narrative literature review: This type of review involves a comprehensive summary and critical analysis of the available literature on a particular topic or research question. It is often used as an introductory section of a research paper. Systematic literature review: This is a rigorous and ...
Rapid review. Assessment of what is already known about a policy or practice issue, by using systematic review methods to search and critically appraise existing research. Completeness of searching determined by time constraints. Time-limited formal quality assessment. Typically narrative and tabular.
Keep it brief. The methods section should be succinct but include all the noteworthy information. This can be a difficult balance to achieve. A useful strategy is to aim for a brief description that signposts the reader to a separate section or sections of supporting information. This could include datasets, a flowchart to show what happened to ...
This paper draws input from a study that employed a systematic literature review as its main source of data. A systematic review can be explained as a research method and process for identifying ...
The Literature Review will place your research in context. It will help you and your readers: Locate patterns, relationships, connections, agreements, disagreements, & gaps in understanding. Identify methodological and theoretical foundations. Identify landmark and exemplary works. Situate your voice in a broader conversation with other writers ...
Sometimes in your literature review, you might need to discuss and evaluate relevant research methodologies in order to justify your own choice of research methodology. When searching for literature on research methodologies it is important to search across a range of sources. No single information source will supply all that you need.
In a literature review, using a single study or fact to "prove" an argument right or wrong is often a signal to the person reading your literature review (usually your professor) that you may not have appreciated the limitations of that study or its place in the broader literature on the topic. ... Advances in Methods and Practices in ...
Purpose: This article aims to critically analyse current literature that explores nurses' perspectives concerning the use of chemical restraints amongst people with dementia. It also aims to consolidate existing knowledge and generate a foundation for further research. Methods: This literature review followed the 12-step approach by Kable et al. A total of 17 articles were included following ...
The systematic literature review employed statistical methods to measure effect sizes and employed traditional univariate systematic literature review to synthesize the results. A table summarizing the literature that met the inclusion criteria was created to ensure transparency and clarity in the data coding process.
Methods. A targeted literature review was conducted. Literature databases (MEDLINE, Embase, and Cochrane reviews) were searched for studies describing clinical practice and treatment outcomes for oral treprostinil and selexipag globally, published in English (2012 to 2022). Electronic searches were supplemented by manual-searches of targeted ...
We investigated publicly available online resources including published literature in Web of Science, PubMed, Google Scholar, and Wanfang database by searching keywords "KBGS", "ANKRD11", "Short stature" and "Intellectual disability" as well as genetic testing records in ClinVar database between July 2011 and March 2024.
This literature review discusses specific challenges associated with drought risk assessments (DRA) within agricultural systems, particularly concerning the justification for indicator selection, aggregation methods, and DRA-informed adaptation strategies. ... DRA-related validation methods being conducted in the literature can be presented in ...
A Review of the Literature on Green Logistics in Supply Chain Management Eydi et al. [ 13 ] investigated a multiple-period multi-echelon closed-loop supply chain network problem for product collection and shipment to determine an optimum number of facilities and their locations as well as the transportation modes.